
- Категория: Блог
- Просмотров: 320
Аннотация
Дистанционное зондирование является полезным инструментом для мониторинга пространственно-временных изменений морфологического и физиологического состояния сельскохозяйственных культур и поддержки методов точного земледелия. По сравнению с мультиспектральной визуализацией гиперспектральная визуализация является более продвинутой технологией, которая способна получать подробную спектральную характеристику целевых объектов. Из-за ограниченной доступности за пределами научного сообщества гиперспектральные изображения не получили широкого распространения в точном земледелии. В последние годы были разработаны различные мини-размерные и недорогие бортовые гиперспектральные датчики (например, CHNSpec FS60C, Headwall Micro-Hyperspec, Cubert UHD 185-Firefly), а также были или будут запущены усовершенствованные космические гиперспектральные датчики (например, PRISMA, DESIS, EnMAP, HyspIRI). Гиперспектральная визуализация становится все более широко доступной для сельскохозяйственных приложений. Между тем, получение, обработка и анализ гиперспектральных изображений по-прежнему остаются сложной темой для исследований (например, большой объем данных, высокая размерность данных и сложный анализ информации).
Поэтому полезно провести тщательный и глубокий обзор технологии гиперспектральной визуализации (например, различных платформ и датчиков), методов, доступных для обработки и анализа гиперспектральной информации, и последних достижений гиперспектральной визуализации в сельскохозяйственных приложениях. Таким образом, были рассмотрены публикации за последние 30 лет по технологии гиперспектральной визуализации и ее применению в сельском хозяйстве. Были обсуждены платформы визуализации и датчики, а также аналитические методы, используемые в литературе. Также были оценены характеристики гиперспектральной визуализации для различных приложений (например, картирование биофизических и биохимических свойств сельскохозяйственных культур, характеристики почвы и классификация сельскохозяйственных культур). Этот обзор призван помочь исследователям и практикам в области сельского хозяйства лучше понять сильные стороны и ограничения гиперспектральной визуализации для сельскохозяйственных приложений и способствовать принятию этой ценной технологии. Также представлены рекомендации по будущим исследованиям гиперспектральной визуализации для точного земледелия.
Ключевые слова:
точное земледелие; дистанционное зондирование; гиперспектральная съемка; платформы и датчики; аналитические методы; свойства сельскохозяйственных культур; характеристики почвы; классификация сельскохозяйственных признаков
ъ 1. Введение
Глобальный сельскохозяйственный сектор сталкивается с растущими проблемами, вызванными рядом стрессовых факторов, включая быстрорастущее население, истощение природных ресурсов, загрязнение окружающей среды, болезни сельскохозяйственных культур и изменение климата. Точное земледелие является перспективным подходом к решению этих проблем путем улучшения методов ведения сельского хозяйства, например, адаптивных ресурсов (например, воды и удобрений), гарантированных результатов (например, урожайности и биомассы) и снижения воздействия на окружающую среду. Дистанционное зондирование способно определять внутриполевые различия почв и сельскохозяйственных культур и предоставлять полезную информацию для методов управления, специфичных для участка [1,2]. Существует два типа технологий дистанционного зондирования с учетом источника энергии: пассивное (например, оптическое) и активное дистанционное зондирование (например, лидар и радар). Пассивное оптическое дистанционное зондирование обычно далее делится на две группы на основе спектрального разрешения датчиков: многоспектральное и гиперспектральное дистанционное зондирование [3]. Мультиспектральная визуализация облегчается путем сбора спектральных сигналов в нескольких дискретных полосах, каждая из которых охватывает широкий спектральный диапазон от десятков до сотен нанометров. Напротив, гиперспектральная визуализация обнаруживает спектральные сигналы в серии непрерывных каналов с узкой спектральной полосой пропускания (например, обычно ниже 10 нм); поэтому она может захватывать мелкомасштабные спектральные характеристики целей, которые в противном случае могли бы быть скомпрометированы [4].
Мультиспектральные изображения (например, Landsat, Sentinel 2 и SPOT) широко используются в сельскохозяйственных исследованиях для получения различных характеристик сельскохозяйственных культур и почв, таких как содержание хлорофилла в сельскохозяйственных культурах, биомасса, урожайность и деградация почвы [5,6,7,8,9,10]. Однако из-за ограничений в спектральном разрешении точность получаемых переменных часто ограничена, и ранние сигналы стресса сельскохозяйственных культур (например, дефицит питательных веществ, болезни сельскохозяйственных культур) не могут быть эффективно обнаружены своевременно [11]. Гиперспектральные изображения (например, CHNspec, Hyperion, CASI и Headwall Micro-Hyperspec) с сотнями каналов могут захватывать более подробные спектральные отклики; следовательно, они более способны обнаруживать тонкие изменения напочвенного покрова и их изменения с течением времени. Таким образом, гиперспектральные изображения могут использоваться для решения вышеупомянутых проблем и способствовать более точному и своевременному обнаружению физиологического состояния сельскохозяйственных культур [12,13]. Предыдущие исследования также продемонстрировали превосходную производительность гиперспектральных изображений по сравнению с мультиспектральными при мониторинге свойств растительности, таких как оценка индекса площади листьев (LAI) [14], различение типов сельскохозяйственных культур [15], извлечение биомассы сельскохозяйственных культур [16] и оценка содержания азота в листьях [17]. Несмотря на свою выдающуюся производительность, гиперспектральная съемка сравнительно меньше использовалась в оперативных сельскохозяйственных приложениях в последние несколько десятилетий из-за высокой стоимости датчиков и миссий по получению изображений, а также различных технических проблем (например, низкое отношение сигнал/шум и большой объем данных)[18,19,20,21]. Хотя данные гиперспектрального отражения на основе наземных данных можно быстро измерить с помощью спектрорадиометра (например,ASD Field Spec, Analytical Spectral Devices Inc., Боулдер, Колорадо, США) и они широко использовались для наблюдения за спектральными особенностями на уровне полога и листьев [22,23,24], такие наземные измерения ограничены несколькими полями и не могут фиксировать пространственную изменчивость на больших площадях. Напротив, гиперспектральные датчики изображений более удобны для получения пространственной изменчивости спектральной информации по региону.
В последние годы был разработан широкий спектр миниатюрных и недорогих гиперспектральных датчиков, которые доступны для коммерческого использования, например, FS60C (CHNSpec Technology, Чжэцзян, Китай), Micro- и Nano-Hyperspec (Headwall Photonics Inc., Бостон, Массачусетс, США), HySpexVNIR (HySpex, Skedsmo, Скьеттен, Норвегия) и FireflEYE (Cubert GmbH, Ульм, Германия) [11,25]. Эти датчики могут быть установлены на пилотируемых или беспилотных воздушных платформах (например, самолетах, вертолетах и беспилотных летательных аппаратах (БПЛА)) для получения гиперспектральных изображений и поддержки различных миссий мониторинга [13,26,27]. Кроме того, недавно были запущены новые космические гиперспектральные датчики, такие как DESIS, запущенный в 2018 году [28] и PRISMA, запущенный в 2019 году [29]—или будут запущены в ближайшие несколько лет, например EnMAP, запуск которого запланирован на 2020 год [30,31]. В целом, все больше и больше гиперспектральных изображений с воздуха или из космоса становятся доступными, что открывает беспрецедентные возможности для лучшего мониторинга наземных целей, особенно для лучшего изучения изменчивости сельскохозяйственных культур и почв и поддержки точного земледелия. Поэтому был проведен поиск литературы, чтобы выяснить, были ли опубликованы дополнительные исследования по использованию гиперспектральных изображений в сельскохозяйственных целях в последние годы. Для проведения поиска литературы по темам или ключевым словам, включая гиперспектральный, визуализация, сельское хозяйство или фермерство, и публикации за 30-летний период времени (с 1990 по 2020 год), использовались как Web of Science, так и Google Scholar. Результаты поиска были дополнительно проверены, чтобы гарантировать, что каждая публикация попадает в область гиперспектральных изображений для сельскохозяйственных приложений. Было обнаружено, что в последние годы растет число публикаций, в которых гиперспектральные изображения использовались для сельскохозяйственных приложений (рисунок 1). За последнее десятилетие было опубликовано существенно больше исследований (например, 245 статей опубликовано в 2011–2020 годах), чем за предыдущее десятилетие (например, 97 статей опубликовано в 2001–2010 годах).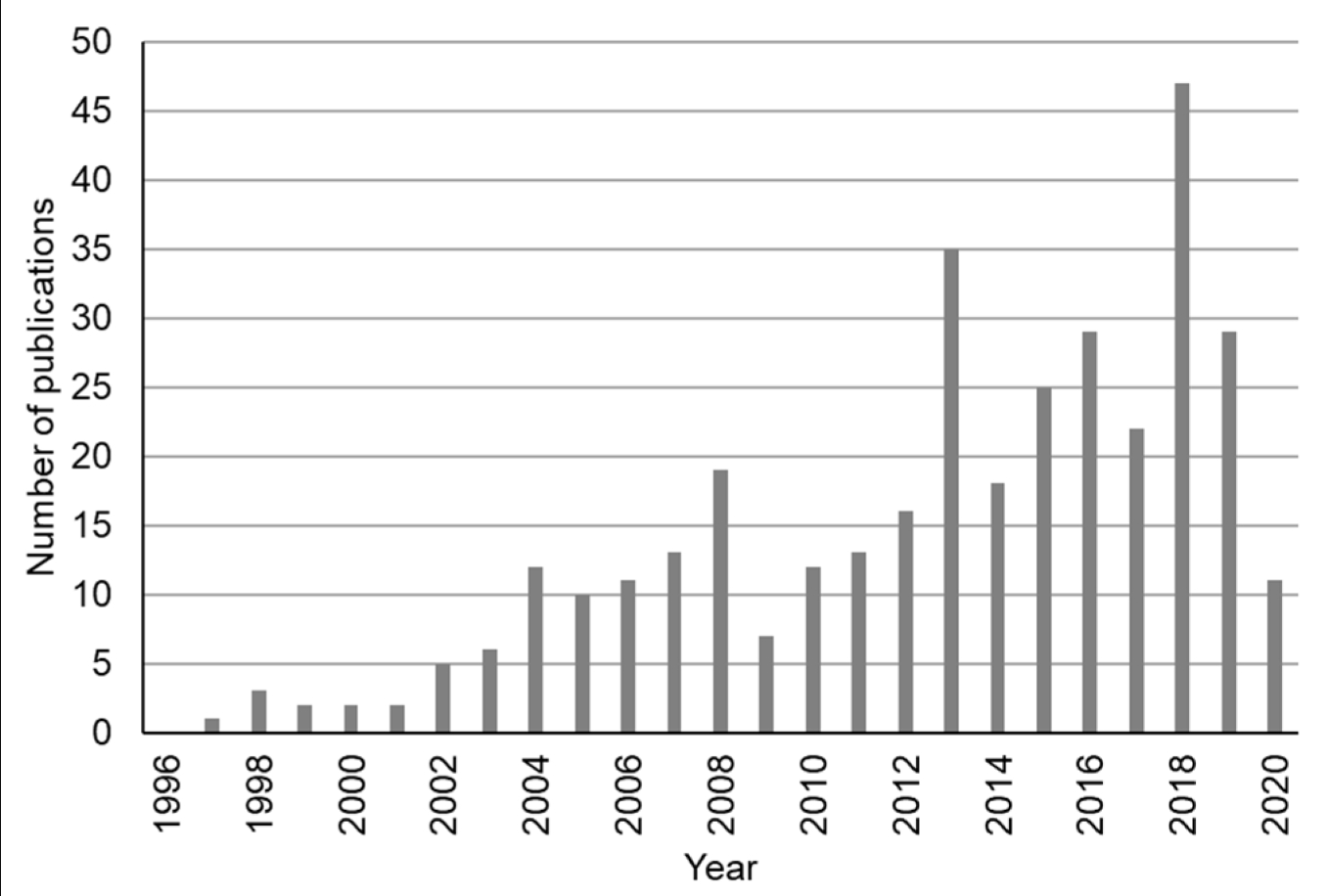
Этот обзор призван сосредоточиться на получении, обработке и анализе гиперспектральных изображений для различных сельскохозяйственных приложений. Обзор организован по следующим основным аспектам: (1) платформы и датчики гиперспектральной визуализации, (2) методы обработки и анализа гиперспектральных изображений и (3) гиперспектральные приложения в сельском хозяйстве (таблица 1). Что касается платформ визуализации, использовались различные типы, включая спутники, самолеты, вертолеты, БПЛА с фиксированным крылом, многороторные БПЛА и платформы ближнего действия (например, наземные или лабораторные). Эти платформы получают изображения с различным пространственным покрытием, пространственным разрешением, временным разрешением, эксплуатационной сложностью и стоимостью миссии. Будет полезно суммировать различные платформы с точки зрения этих характеристик, чтобы поддержать выбор подходящей платформы(-ок) для различных целей мониторинга. После получения необработанных гиперспектральных изображений предварительная обработка является шагом для получения точной спектральной информации. Несколько процедур должны быть выполнены во время предварительной обработки (обычно реализуемой в специализированном программном обеспечении дистанционного зондирования), включая радиометрическую калибровку, спектральную коррекцию, атмосферную коррекцию и геометрическую коррекцию. Хотя это стандартные этапы обработки для большинства спутниковых изображений, их все еще может быть сложно выполнить на многих гиперспектральных изображениях с воздуха из-за различных технических проблем (например, требование высокоточных сигналов Глобальной системы позиционирования (GPS) для правильной геометрической коррекции, измерение солнечного излучения в реальном времени для точной спектральной коррекции). Не существует стандартизированных протоколов для всех датчиков из-за ограниченной доступности гиперспектральных изображений в прошлом и того факта, что новые мини-размеры и низко-гиперспектральные датчики cost на рынке представлены разными производителями с различными конфигурациями датчиков. В предыдущих исследованиях использовались различные подходы для решения этих проблем [12,19,32,33]. Поэтому важно пересмотреть эти подходы, чтобы поддержать других исследователей в более точной и эффективной обработке гиперспектральных изображений. После предварительной обработки, такой как калибровка и коррекция, можно выполнить извлечение спектральной информации (например, выбор полосы и уменьшение размерности) для дальнейшего улучшения удобства использования гиперспектрального изображения. Методы для этих процедур рассматриваются в этом исследовании.
При использовании предварительно обработанных гиперспектральных изображений требуется надежный и эффективный аналитический метод для анализа огромного количества информации, содержащейся в изображениях (например, спектральные, пространственные и текстурные характеристики), и извлечения целевых свойств (например, характеристики сельскохозяйственных культур и почвы). В предыдущих исследованиях использовался набор аналитических методов, включая эмпирическую регрессию (например, линейную регрессию, частичную регрессию наименьших квадратов (PLSR) и многомерную переменная регрессия (MLR)), моделирование переноса излучения (RTM, например, PROSPECT и PROSAIL), машинное обучение (например, случайный лес (RF)) и глубокое обучение (например, сверточная нейронная сеть (CNN)) [34,35,36,37]. Эти методы были разработаны на основе разных теорий и имеют разную операционную сложность, вычислительную эффективность и точность производительности. Поэтому важно рассмотреть сильные и слабые стороны этих методов и выбрать подходящий для конкретных исследовательских целей. Используя гиперспектральную информацию, исследователи изучили широкий спектр сельскохозяйственных характеристик. Некоторые популярные из них включают содержание воды в культурах, LAI, содержание хлорофилла и азота, вредителей и болезни, высоту растений, фенологическую информацию, влажность почвы и содержание органических веществ в почве [11,38]. Также будет полезно рассмотреть характеристики гиперспектральной визуализации в этих исследованиях и дополнительно изучить потенциал этой технологии для мониторинга других сельскохозяйственных характеристик. Наконец, обсуждаются проблемы использования гиперспектральных изображений для точного земледелия, а также будущие направления исследований. Несколько предыдущих обзорных статей в некоторой степени обсуждали некоторые из этих тем [11,38,39]. Более подробная информация и вклад этого обзора будут обсуждаться в каждом конкретном разделе. В целом, этот обзор направлен на изучение основных процедур сбора и использования гиперспектральных изображений для различных сельскохозяйственных приложений, чтобы глубже понять сильные стороны и ограничения гиперспектральной технологии и способствовать более быстрому внедрению этой ценной технологии в точном земледелии..
2. Платформы и датчики гиперспектральной визуализации
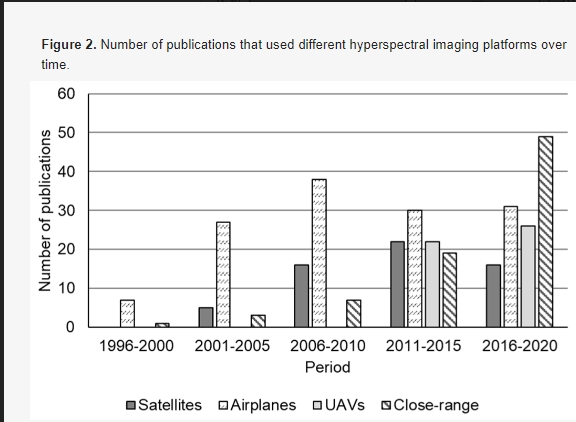
Гиперспектральные датчики могут быть установлены на различных платформах, таких как спутники, самолеты, беспилотные летательные аппараты и платформы ближнего действия, для получения изображений с различным пространственным и временным разрешением. Платформы, используемые в литературе, были идентифицированы и обобщены за годы публикации с целью найти, если таковые имеются, платформы, которые использовались чаще всего в определенный период времени, и результаты показаны на рисунке 2. Самолеты были наиболее широко используемыми платформами для гиперспектральной съемки в сельском хозяйстве (рисунок 2). Примерно 30 статей, в которых использовались самолеты, публиковались каждые пять лет, начиная с 2001 года (например, 27 публикаций в 2001–2005 годах и 38 в 2006–2010 годах). Для сравнения, гиперспектральная съемка на основе спутников использовалась реже; примерно 20 или менее статей были опубликованы за все пятилетние периоды. БПЛА являются популярными платформами для дистанционного зондирования и широко использовались в последнее десятилетие для гиперспектральной визуализации в сельском хозяйстве (например, более 20 публикаций в 2011–2015 и 2016–2020 годах). Платформы ближнего действия наиболее широко использовались в последние пять лет (т. е. 2016–2020 годы), с 49 публикациями (рисунок 2). Обзор в этом разделе структурирован на основе различных платформ, включая спутники, самолеты, БПЛА и платформы ближнего действия. В отличие от предыдущих статей, рассматривающих гиперспектральные платформы [20,38,39], обзор в этом разделе больше фокусируется на последних достижениях платформ визуализации (например, БПЛА, вертолеты и ближний диапазон) и их приложениях в точном земледелии (например, классификация сорняков, мелкомасштабная оценка здоровья сельскохозяйственных культур, вредителей и болезней).
2.1. Спутниковая гиперспектральная съемка
По сравнению с большим количеством спутниковых мультиспектральных датчиков (например, Landsat, SPOT, WorldView, QuickBird, Sentinel-2), гиперспектральных датчиков значительно меньше. EO-1 Hyperion, PROBA-CHRIS и TianGong-1 [40] — вот несколько примеров доступных спутниковых гиперспектральных датчиков [20]. EO-1 Hyperion — наиболее широко используемый спутниковый гиперспектральный датчик для сельского хозяйства (например, более 40 публикаций). Он собирает данные в видимом, ближнем инфракрасном и коротковолновом инфракрасном диапазонах со спектральным разрешением 10 нм и пространственным разрешением 30 м. Дополнительные характеристики сенсора EO-1 Hyperion приведены в Таблице 2. Сенсор работал с 2000 по 2017 год, что соответствует периоду с большим количеством публикаций с использованием спутниковой гиперспектральной съемки (например, с 2006 по 2020 год на Рисунке 2). Использование данных Hyperion было описано в различных сельскохозяйственных исследованиях для мониторинга различных свойств сельскохозяйственных культур и почвы, включая обнаружение заболеваний сельскохозяйственных культур [41,42], оценку свойств сельскохозяйственных культур (например, хлорофилла, LAI, биомассы) [43,44,45], оценку остатков сельскохозяйственных культур [46,47], классификацию типов сельскохозяйственных культур [48] и исследование свойств почвы [49,50]. Несколько представленных работ включают Wu et al. [45], которые оценили содержание хлорофилла в растительности и LAI на смешанном сельскохозяйственном поле с использованием данных Hyperion и оценили спектральные диапазоны, которые чувствительны к этим свойствам растительности. Camacho Velasco et al. [48] использовали гиперспектральные изображения Hyperion и различные алгоритмы классификации (например, картограф спектрального угла и адаптивный оценщик когерентности) для идентификации пяти типов сельскохозяйственных культур (например, масличная пальма, каучук, трава для выпаса скота, цитрусовые и сахарный тростник) в Колумбии. Гомес и др. [49] предсказали органический углерод почвы (SOC), используя как данные спектрорадиометра, так и гиперспектральное изображение Hyperion, и они обнаружили, что использование данных Hyperion привело к более низкой точности по сравнению с результатами, полученными с помощью данных спектрорадиометра.
Также были проведены исследования для сравнения производительности гиперспектральных изображений Hyperion с многоспектральными изображениями для оценки свойств сельскохозяйственных культур или классификации типов сельскохозяйственных культур. Например, Мариотто и др. [15] сравнили гиперспектральные изображения Hyperion с многоспектральными изображениями Landsat для оценки урожайности сельскохозяйственных культур и классификации типов сельскохозяйственных культур. Авторы сообщили о более высоких производительностях использования гиперспектральных изображений, чем использования изображений Landsat для обеих исследовательских целей. Аналогичным образом, Бостан и др. [51] сравнили гиперспектральные изображения Hyperion с многоспектральными изображениями Landsat для классификации сельскохозяйственных культур и также обнаружили, что более высокая точность классификации может быть достигнута с помощью гиперспектральных изображений.
PROBA-CHRIS — еще один широко используемый спутниковый гиперспектральный датчик, который был запущен в 2001 году. Конкретные исследования, такие как Verger et al. [57] использовали данные PROBA-CHRIS для получения LAI, доли растительного покрова (fCover) и доли поглощенной фотосинтетически активной радиации (FAPAR) на сельскохозяйственном поле. Энтони и др. [58] определили три стадии роста пшеницы с помощью многоракурсных изображений PROBA-CHRIS и нашли оптимальные углы обзора для идентификации. Каса и др. [59] оценили производительность данных бортового мультиспектрального инфракрасного видимого спектрометра (MIVIS) и космических данных PROBA-CHRIS для исследования текстуры почвы и обнаружили, что эти два типа данных имеют схожие характеристики, хотя данные PROBA-CHRIS имеют более низкое пространственное разрешение.
Существует несколько других гиперспектральных сенсоров на основе спутников, которые обычно не используются в сельскохозяйственной среде. Например, Hyperspectral Imager (HySI) — это гиперспектральный сенсор, установленный на индийском микроспутнике IMS-1, запущенном в 2008 году [60]. Он собирает спектральные сигналы в диапазоне 400–950 нм с пространственным разрешением 550 м в надире [61]. Изображения HySI использовались для картирования различных сельскохозяйственных характеристик, таких как влажность почвы и засоленности почвы [62]. Они также использовались для классификации сельскохозяйственных культур [63]. Однако эти данные не получили широкого распространения в точном земледелии, что, вероятно, связано с низким пространственным разрешением и ограниченной доступностью данных. Hyperspectral Imager for the Coastal Ocean (HICO) — это еще один космический гиперспектральный сенсор, который делает снимки в спектральном диапазоне от 380 до 960 нм с пространственным разрешением 90 м [64]. Этот датчик был в основном разработан для сбора данных в прибрежной зоне океана и эксплуатировался с 2009 по 2015 год.
В последние годы было запущено или запланировано к запуску в ближайшие несколько лет несколько космических гиперспектральных датчиков. Например, спектрометр для зондирования Земли (DESIS) Германского аэрокосмического центра (DLR), гиперспектральный датчик, установленный на Международной космической станции, был запущен в 2018 году [65]. Этот датчик получает изображения в диапазоне от 400 до 1000 нм со спектральным разрешением 2,5 нм и пространственным разрешением 30 м. Hyperspectral Imager Suite (HISUI) — японский гиперспектральный датчик, который также находится на борту Международной космической станции [66]. Он был запущен в 2019 году и собирает данные в диапазоне от 400 до 2500 нм с пространственным разрешением 20 м и временным разрешением от 2 до 60 дней [20]. Hyperspectral Precursor and Application Mission (PRISMA) — итальянская гиперспектральная миссия с датчиком, запущенным в марте 2019 года. Ее спектральное разрешение составляет 12 нм в диапазоне 400–2500 нм (~250 полос в видимом и коротковолновом инфракрасном диапазонах). Ее гиперспектральные изображения имеют пространственное разрешение 30 и 5 м для панхроматического диапазона [67]. Environmental Mapping and Analysis Program (EnMAP) — немецкая гиперспектральная спутниковая миссия, которая все еще находится на стадии разработки и производства [68]. Датчик EnMAP будет собирать данные в видимом и коротковолновом инфракрасном диапазонах с пространственным разрешением 30 м. Его запуск запланирован на 2020 год. Spaceborne Hyperspectral Applicative Land and Ocean Mission (SHALOM) — совместная миссия израильских и итальянских космических агентств, и запуск спутника запланирован на 2022 год [69]. Этот датчик будет собирать гиперспектральные изображения с пространственным разрешением 10 м в спектральном диапазоне 400–2500 нм и панхроматические изображения с пространственным разрешением 2,5 м [70]. HyspIRI — еще одна гиперспектральная миссия, которая также находится на стадии изучения [71]. Этот датчик будет собирать данные в диапазоне от 380 до 2500 нм с интервалом 10 нм и пространственным разрешением 60 м.
Хотя фактические данные PRISMA, EnMAP и HyspIRI пока недоступны, исследователи смоделировали изображения с использованием других данных и протестировали производительность смоделированных изображений для исследования различных характеристик растительности и почвы. Например, Малек и др. [72], Зигманн и др. [73] и Лохерер и др. [74] смоделировали изображения EnMAP с использованием различных изображений, полученных с воздуха или из космоса, и применили смоделированные изображения для исследования различных свойств сельскохозяйственных культур и почвы. Бахманн и др. [75] создали изображение с помощью инструмента сквозного моделирования EnMAP и изучили неопределенности, связанные со спектральной и радиометрической калибровкой. Кастальди и др. [76] смоделировали данные четырех текущих (EO-1 ALI и Hyperion, Landsat 8 Operational Land Imager (OLI), Sentinel-2 MultiSpectral Instrument (MSI)) и трех будущих (EnMAP, PRISMA и HyspIRI) датчиков, используя библиотеку спектров почвы, и сравнили их производительность для оценки свойств почвы. Кастальди и др. [77] использовали данные PRISMA, которые были смоделированы с помощью спектральных данных, измеренных в лаборатории, для оценки содержания глины и попытались уменьшить влияние влажности почвы на оценку содержания глины.
Предыдущие исследования подтвердили хорошую производительность спутниковых гиперспектральных датчиков для изучения сельскохозяйственных особенностей; однако, несколько факторов могут потенциально повлиять на широкое применение этих данных в точном земледелии, включая пространственное разрешение, временное разрешение и качество данных. Обнаружение и мониторинг многих сельскохозяйственных особенностей, таких как болезни сельскохозяйственных культур, заражение вредителями и состояние питательных веществ, требуют высокого пространственного и временного разрешения. Большинство спутниковых гиперспектральных датчиков имеют среднее пространственное разрешение, например 17 или 36 м для PROBA-CHRIS; 30 м для Hyperion, PRISMA и EnMAP, DESIS; и 60 м для HyspIRI. Предыдущие исследования показали, что такое пространственное разрешение недостаточно для приложений точного земледелия [20,49]. Чтобы преодолеть такие ограничения, исследователи попытались повысить резкость гиперспектральных изображений, стремясь улучшить пространственное разрешение [73,78,79,80]. Loncan et al. [81] также рассмотрели различные методы паншарпенинга для создания гиперспектральных изображений с высоким пространственным разрешением.
Временное разрешение является еще одним фактором, который может потенциально ограничить применение спутниковых гиперспектральных изображений в точном сельском хозяйстве. Большинство спутниковых датчиков имеют длительный цикл повторного обзора (например, обычно около двух недель), и, таким образом, ранние сигналы стресса сельскохозяйственных культур (например, болезни и вредители) могут быть пропущены. Это ограничение может еще больше усугубляться неблагоприятными погодными условиями (например, загрязнением облаков). Наконец, низкое качество данных также является проблемой, которая может повлиять на производительность спутниковых гиперспектральных изображений для исследования сельскохозяйственных объектов. Низкое отношение сигнал/шум является хорошо известной проблемой данных Hyperion (например, в коротковолновом инфракрасном диапазоне (SWIR)), что повлияло на точность получения различных сельскохозяйственных объектов [20]. Например, Аснер и Хайдебрехт [82], Гомес и др. [49] и Венг и др. [83] обнаружили, что низкое отношение сигнал/шум повлияло на точность оценки нефотосинтетической растительности и почвенного покрова, органического вещества почвы и солености почвы соответственно. Ожидается, что будущие гиперспектральные миссии на основе спутников решат проблему качества данных.
2.2. Гиперспектральная съемка с борта самолета
Гиперспектральная съемка с борта самолета широко использовалась для сбора гиперспектральных изображений для различных целей мониторинга (например, для сельского или лесного хозяйства). Первым гиперспектральным датчиком был бортовой видимый/инфракрасный спектрометр (AVIRIS), который был разработан и использован в 1987 году [84]. Он собирает спектральные сигналы в 224 полосах в видимом диапазоне до SWIR (таблица 2). Исследователи использовали данные AVIRIS, чтобы помочь понять широкий спектр сельскохозяйственных особенностей, таких как исследование свойств растительности (например, урожайность, LAI, хлорофилл и содержание воды) [85,86,87,88], анализ свойств почвы [89], оценка здоровья сельскохозяйственных культур или выявление заражения вредителями [90,91,92] и картирование посевных площадей или методов обработки почвы [93,94].
Помимо AVIRIS, широко используются также бортовые гиперспектральные датчики Compact Airborne Spectrographic Imager (CASI), Hyperspectral Mapper (HyMap) и AISA Eagle (таблица 2). Например, изображения CASI использовались для оценки содержания хлорофилла в культурах [95], исследования доли покрытия культурами [96], классификации сорняков [97] и разграничения зон управления [2]. Изображения HyMap применялись для изучения биофизических и биохимических переменных культур (например, LAI, содержание хлорофилла и воды) [98,99,100], обнаружения сигналов стресса растений [101] и исследования пространственных закономерностей SOC [102]. Что касается изображений AISA Eagle, Рю и др. [35] и Силия и др. [103] использовали эти данные для оценки содержания азота в культурах, а Амбрус и др. [104] использовали их для оценки биомассы.
Несколько других гиперспектральных датчиков с борта самолета также использовались в предыдущих исследованиях. Например, изображения AVIS использовались для исследования ряда характеристик растительности (например, биомассы и хлорофилла) [105], гиперспектральные изображения Probe-1 использовались для исследования остатков урожая [106], гиперспектральные изображения RDACS-H4 использовались для обнаружения болезней урожая [34], гиперспектральный датчик AHS-160 использовался для картирования SOC [107], датчик SWIR Hyper Spectral Imaging (HSI) использовался для оценки влажности почвы [108], гиперспектральный сканер Pushbroom (PHI) использовался для оценки LAI озимой пшеницы [109], а данные эксперимента с призмой с борта самолета (APEX) использовались для изучения связи между SOC на пахотных землях и спектральными сигналами [110].
Большинство вышеупомянутых гиперспектральных изображений с воздуха были получены самолетами на средней и большой высоте (например, высота 1–4 км для CASI, 20 км для AVIRIS), и полученные изображения обычно имеют высокое или среднее пространственное разрешение, например, 4 м для изображений CASI, 5 м для HyMap и 20 м для AVIRIS [111,112,113]. Такое пространственное разрешение подходит для картирования многих особенностей сельскохозяйственных культур и почв. Однако получение изображений обычно необходимо планировать за несколько месяцев или даже лет, а полеты обходятся дорого [19]. Кроме того, для некоторых конкретных приложений, таких как исследование особенностей на уровне видов или сообществ (например, идентификация сорняков или ранний сигнал о заболевании сельскохозяйственных культур), предпочтительны изображения с очень высоким пространственным разрешением (например, субметровое) [114,115]. Кроме того, из-за нестабильной природы самолетов как платформ для получения изображений, потребуется карданный подвес или высокоточный инерциальный измерительный блок (IMU) для компенсации изменения ориентации самолетов или записи информации об ориентации для последующей коррекции изображения соответственно. Эти факторы ограничили полное применение гиперспектральной съемки с воздуха в точном сельском хозяйстве. Пилотируемые вертолеты также использовались в качестве платформ для гиперспектральной съемки и исследования особенностей растительности [27,116]. Вертолеты имеют более гибкие высоты полета (например, 100 м–2 км), чем самолеты, и способны получать изображения с высоким пространственным разрешением (например, субметровые) на больших площадях. Для задачи получения изображений, как правило, требуется авиационная компания с пилотируемым вертолетом, что требует дополнительной финансовой поддержки и далеко идущего предварительного планирования.
2.3. Гиперспектральная съемка с помощью БПЛА
В последние годы БПЛА стали популярной платформой для получения данных дистанционного зондирования, особенно для мультиспектральной съемки с использованием цифровых камер или мультиспектральных датчиков. С ростом доступности легких гиперспектральных датчиков исследователи экспериментировали с установкой этих датчиков на БПЛА для получения гиперспектральных изображений с высоким пространственным разрешением [19,117]. В предыдущих исследованиях использовались различные типы БПЛА, включая многороторные, вертолеты и с фиксированным крылом (рисунок 3). По сравнению с пилотируемыми самолетами и вертолетами, БПЛА способны получать изображения с высоким пространственным разрешением с гораздо меньшей стоимостью и обладают высокой гибкостью с точки зрения планирования полетной миссии [118]. Несколько конкретных сельскохозяйственных применений БПЛА -
Результаты гиперспектральной визуализации приведены в таблице 3.

Различные легкие гиперспектральные датчики были разработаны в последние годы и могут быть установлены на БПЛА. Примерами датчиков являются широко используемые CHNSpec FS60C, Headwall Micro- и Nano-Hyperspec VNIR [12,13,26,128], UHD 185-Firefly [53,130], датчик PIKA II [19,32] и HySpex VNIR [25,131]. Эти гиперспектральные датчики содержат более 100 полос в видимом-ближнем инфракрасном спектральном диапазоне (таблица 2). Эти датчики небольшие и компактные (1-2 кг), поэтому их можно быстро развернуть на различных пилотируемых или беспилотных платформах дистанционного зондирования. Предыдущие исследования, проведенные Адао и др. [11] и Лодхи и др. [52], также сравнили и обобщили различные легкие гиперспектральные датчики.
При применении гиперспектральной съемки с использованием БПЛА необходимо учитывать большое количество факторов, начиная от настройки сенсора и сбора данных и заканчивая обработкой изображений. Саари и др. [122] проверили осуществимость системы гиперспектральной съемки с использованием БПЛА для сельскохозяйственных и лесных применений и обсудили несколько проблем, связанных с технологией съемки (например, требования к оборудованию и системные настройки). Аасен и др. [132] сосредоточились на калибровке изображений, полученных с помощью кадрового сенсора, и обсудили несколько проблем, связанных с использованием гиперспектральной съемки с использованием БПЛА для исследования растительности и сельскохозяйственных культур (например, полезная нагрузка БПЛА, отношение сигнал/шум и спектральная калибровка). Хабиб и др. [120] попытались выполнить орторектификацию гиперспектральной съемки с использованием беспилотного летательного аппарата с помощью кадровых изображений RGB над сельскохозяйственным полем. Адао и др. [11] рассмотрели применение гиперспектральных изображений на основе БПЛА в сельском и лесном хозяйстве и перечислили несколько гиперспектральных датчиков, которые могут быть установлены на БПЛА. Авторы также обсудили несколько проблем при сборе и анализе гиперспектральных изображений на основе БПЛА, таких как радиометрический шум, низкое качество геопривязки БПЛА и низкое отношение сигнал/шум.
Гиперспектральная съемка с использованием БПЛА стала более популярной в последние годы; поэтому крайне важно рассмотреть ее сильные и слабые стороны. Чтобы изучить больше возможностей этой технологии, этот раздел обзора не ограничивается только сельскохозяйственными приложениями. Различные типы БПЛА использовались в качестве платформ для гиперспектральной съемки, причем два из них наиболее широко использовались как многороторные [130,133,134] и самолеты с фиксированным крылом [33,120,135]. Медленные полеты на малых высотах предпочтительны для получения гиперспектральных изображений с высоким пространственным разрешением и высоким отношением сигнал/шум. Таким образом, многороторные самолеты более конкурентоспособны, чем самолеты с фиксированным крылом для гиперспектральной съемки с точки зрения эксплуатации полета. В частности, многороторные самолеты обеспечивают низкую высоту полета, гибкую скорость полета и вертикальный взлет и посадку, в то время как для самолета с фиксированным крылом требуются минимальная высота полета, скорость и, иногда, аксессуары для взлета и посадки (например, взлетно-посадочная полоса, пусковая установка и парашют). Система гиперспектральной визуализации, состоящая из гиперспектрального датчика, блока обработки данных, GPS и IMU, имеет значительный вес (например, 1–3 кг), что создает проблемы для грузоподъемности системы БПЛА и срока службы ее батареи. Мультироторы, как правило, питаются от высокопроизводительных батарей (например, LiPo), и большинство из них имеют короткую продолжительность полета (например, менее 20 мин). Продолжительность полета может составлять всего 3 мин [12]. Напротив, многие БПЛА с фиксированным крылом работают на топливе, поэтому имеют гораздо большую продолжительность полета (например, 1–10 ч) [19,135]. Однако эти самолеты с фиксированным крылом в основном большие и тяжелые (например, размах крыльев 5 м и взлетный вес 14 кг) [135], и, таким образом, создают проблемы для летной эксплуатации. Используя БПЛА, исследователям необходимо учитывать SWaP БПЛА (размер, вес и мощность), географическое покрытие, время в воздухе, высоту и другие переменные. В дополнение к проблемам, связанным с созданием системы БПЛА и выполнением полетов, исследователям, вероятно, потребуется подать заявку на получение разрешения на полет от авиационного органа (например, сертификат специальных полетных операций (SFOC) от Transport Canada) и приобрести подходящую страховку для полетов БПЛА [136]. Размер и вес БПЛА являются важными параметрами, которые следует учитывать в этих процессах. Кроме того, БПЛА должны быть видны во время полетных миссий, чтобы пилот мог поддерживать постоянный визуальный контакт с самолетом. Это может создать серьезную проблему при полете над большой территорией, холмистой местностью или лесной местностью.
2.4. Гиперспектральная съемка с близкого расстояния (наземная или лабораторная) Гиперспектральная съемка с близкого расстояния, включая наземную (рисунок 4a–c) или лабораторную (рисунок 4d,e), является новой технологией последних лет, и она способна получать гиперспектральные изображения сверхвысокого пространственного разрешения (например, на уровне см или ниже см) [137,138,139]. Таким образом, эта технология визуализации может использоваться для исследования мелкомасштабных (например, на уровне листьев и полога) особенностей растительности и, таким образом, в значительной степени поддерживает исследование состояния роста сельскохозяйственных культур и обнаружение ранних признаков стресса сельскохозяйственных культур (например, болезни, сорняки или дефицит питания). Датчики устанавливаются на движущихся или статических платформах (например, линейных платформах, лесах или грузовиках), которые могут быть развернуты в помещении или на открытом воздухе для сбора изображений. В качестве источников света в этих платформах используются лампы (например, галогенные) или солнце соответственно.

В целом, платформа гиперспектральной визуализации с близкого расстояния способна получать гиперспектральные изображения сверхвысокого пространственного разрешения, которые имеют решающее значение для исследования мелкомасштабных характеристик сельскохозяйственных культур или почвы. Эти характеристики предоставляют подробную информацию о биофизических и биохимических процессах растений и о том, как растения реагируют на экологические стрессы и заболевания. Однако сбор и обработка изображений также страдают от различных проблем, таких как неинформативная изменчивость, вызванная взаимодействием света со структурой растения (т. е. эффекты освещения), влияние теней и расширение приложений платформы до больших масштабов [141,146]. Дальнейшие исследования в этих областях оправданы.
Подводя итог, различные платформы гиперспектральной визуализации, включая спутники, самолеты, вертолеты, БПЛА и ближний диапазон, имеют различные преимущества и недостатки для применения в точном сельском хозяйстве. Подробные сравнения этих платформ для сельскохозяйственных приложений показаны в Таблице 5. Вкратце, спутниковые системы предоставляют изображения, охватывающие большие площади, но страдают от среднего пространственного разрешения и ограниченной доступности данных (например, ограниченное количество рабочих датчиков и длительное время повторного просмотра). Платформы визуализации на базе самолетов и вертолетов получают данные с подходящим пространственным покрытием и разрешением для большинства сельскохозяйственных приложений. Однако они ограничены высокой стоимостью миссии и проблемами планирования и, таким образом, не подходят для повторного мониторинга. Системы на базе БПЛА способны многократно получать изображения с высоким пространственным разрешением и обладают высокой гибкостью. Однако они могут охватывать только небольшую площадь из-за ограниченного срока службы батареи и авиационных правил. Системы визуализации с близкого расстояния способны получать изображения со сверхвысоким пространственным разрешением, но их можно использовать только на уровне листьев или полога. Поэтому при выборе платформы для конкретного исследовательского проекта следует учитывать следующие факторы: пространственное разрешение, необходимое для исследования, область полета и выносливость полета, вес системы визуализации, грузоподъемность платформы, безопасность и правила полета, эксплуатационная гибкость и стоимость.
3. Методы обработки и анализа гиперспектральных изображений
Гиперспектральные изображения, полученные различными платформами и датчиками, обычно предоставляются в необработанном формате (например, цифровые числа), который необходимо предварительно обработать (например, атмосферные, радиометрические и спектральные поправки) для получения точной спектральной информации. После этого можно использовать различные подходы для анализа гиперспектральной информации и исследования различных сельскохозяйственных характеристик (например, свойств сельскохозяйственных культур и почвы). Несколько часто используемых методов включают линейную регрессию, расширенную регрессию (например, PLSR), машинное обучение и глубокое обучение (например, RF, CNN) и моделирование переноса излучения (например, PROSPECT и PROSAIL). Исследователи использовали один или несколько из этих методов для исследования различных сельскохозяйственных характеристик. В этом разделе обзор организован на основе различных методов, используемых в исследованиях.
3.1. Предварительная обработка гиперспектральных изображений
Типичная обработка гиперспектральных изображений включает геометрическую коррекцию, ортотрансформацию, радиометрическую коррекцию и атмосферную коррекцию. Для спутниковых и самолетных гиперспектральных изображений геометрическая и ортотрансформационная коррекция обычно выполняются поставщиками данных, а радиометрическая и атмосферная коррекции могут быть выполнены с помощью стандартных шагов обработки изображений, доступных в программном обеспечении для дистанционного зондирования. Для изображений с БПЛА, напротив, пользователям необходим о выполнить эти шаги обработки и выбрать соответствующие методы обработки и связанные параметры. Например, для выполнения ортотрансформации и геометрической коррекции обычно требуются цифровая модель рельефа (ЦМР) и наземные контрольные точки (GCP) [12]. Если датчик, установленный на БПЛА, основан на сканировании, для этих коррекций потребуется точная информация об ориентации датчика, записанная IMU, а IMU должен быть интегрирован в БПЛА и хорошо откалиброван [12,27]. Пакеты программного обеспечения, обычно используемые в предыдущих исследованиях для выполнения этих коррекций на гиперспектральных изображениях, полученных с помощью БПЛА, включают ENVI (Exelis Visual Information Solutions, Боулдер, Колорадо, США) и PARGE (ReSe Applications Schläpfer, Виль, Швейцария) [12,26,117].
Радиометрическая коррекция проводится для преобразования цифровых чисел изображения в яркость с использованием калибровочных коэффициентов, которые предоставляются производителем сенсора [11]. Эти коэффициенты могут потребовать обновления с течением времени из-за деградации спектральных материалов, используемых для создания гиперспектральных сенсоров. Что касается атмосферной коррекции, хотя БПЛА летают на малых высотах, полученные сигналы по-прежнему субъективны к влиянию различных атмосферных поглощений и рассеяний, таких как поглощение кислорода на 760 нм; поглощение воды около 820, 940, 1140, 1380 и 1880 нм; и поглощение углекислого газа на 2010 и 2060 нм [12,13,26,150]. Поэтому атмосферная коррекция имеет решающее значение для получения спектральной информации хорошего качества. Однако Адао и др. [11] предполагают, что этот процесс можно пропустить, если БПЛА работают близко к земле. Таким образом, применение атмосферной коррекции будет зависеть от конкретных полетных миссий и исследовательских целей (например, высоты полета, если необходимы спектральные полосы, подверженные влиянию атмосферы). Программное обеспечение или методы, обычно используемые в предыдущих исследованиях для выполнения атмосферной коррекции на гиперспектральных изображениях, полученных с помощью БПЛА, включают модель MODTRAN (Spectral Sciences Inc.), ENVI FLAASH (L3Harris Geospatial), PCI Geomatica (PCI Geomatics Corporate), модель SMARTS (Solar Consulting Services) и эмпирическую линейную коррекцию [12,19,27,32,33,116].
Гиперспектральные изображения обычно имеют сотни каналов, и многие из них сильно коррелируют. Поэтому уменьшение размерности также является важной процедурой для рассмотрения при предварительной обработке гиперспектральных изображений. Во многих предыдущих исследованиях с использованием гиперспектральных изображений обсуждались проблемы избыточности данных и использовались различные методы уменьшения размерности. Например, Миглани и др. [151] выполнили анализ главных компонент (PCA) на гиперспектральных изображениях и указали, что 99% информации можно объяснить в первых 10 главных компонентах. Амато и др. [152] обсудили несколько предыдущих методов уменьшения размерности, таких как PCA, минимальная доля шума (MNF) и разложение по сингулярным значениям (SVD), и предложили алгоритм уменьшения размерности, основанный на дискриминантном анализе для контролируемой классификации. Теке и др. [38] рассмотрели несколько методов уменьшения размерности и обобщили их на основе методов преобразования. Тханкабаил и др. [153] обсудили проблемы высокой размерности и перечислили ряд спектральных диапазонов, которые более важны для исследования признаков урожая. Sahoo et al. [4] рассмотрели различные методы снижения размерности, такие как PCA, единообразный дизайн признаков (UMD), вейвлет-преобразования и искусственные нейронные сети (ANN), и обсудили особенности их работы. Wang et al. [154] предложили метод снижения размерности на основе автокодировщика, который является подходом, основанным на глубоком обучении. Из этих различных методов вейвлет-преобразование является одним из наиболее широко используемых для снижения размерности. Этот метод разлагает сигнал на ряд масштабированных версий материнской вейвлет-функции и позволяет варьировать вейвлет на основе информации о частоте для извлечения локализованных признаков (например, локальной спектральной вариации) [155,156]. Он также успешно использовался для слияния изображений, извлечения признаков и классификации изображений [156,157,158].
В дополнение к снижению размерности, анализ чувствительности полосы и выбор полосы также широко использовались в гиперспектральном дистанционном зондировании для уменьшения размера данных путем выбора только полос, чувствительных к интересующему объекту. В предыдущих исследованиях были предложены различные алгоритмы для выбора полосы, такие как быстрый метод на основе градиента объема, который является неконтролируемым методом и последовательно удаляет самую избыточную полосу на основе градиента объема [159], метод на основе выбора подмножества столбцов, который максимизирует объем выбранного подмножества столбцов (т. е. полос) и является устойчивым к шумным полосам [160], и метод выбора заметной полосы на основе ранжирования многообразий, который помещает векторы полос в пространство многообразий и выбирает ранжирование на основе полос, которое может решить проблему ненадлежащего измерения разницы полос [161]. С помощью анализа чувствительности предыдущие исследования определили спектральные полосы, которые чувствительны к различным свойствам сельскохозяйственных культур, например, ~515, ~550, ~570, ~670, 700–740, ~800 и ~855 нм для исследования содержания хлорофилла; ~405, ~515, ~570, ~705 и ~720 нм для оценки состояния азота; ~970, ~1180, ~1245, ~1450 и ~1950 нм для оценки содержания воды; ~682, ~855, ~910, ~970, ~1075, ~1245, ~1518, ~1725 и ~2260 нм для оценки биомассы; и ~550, ~682, ~855, ~1075, ~1180, ~1450 и ~1725 нм для классификации сельскохозяйственных культур [36,44,153,162]. В целом, предварительная обработка является важным шагом для улучшения качества гиперспектральных изображений и подготовки к дальнейшему анализу данных. После предварительной обработки аналитические методы, которые будут обсуждаться ниже, могут быть использованы для анализа гиперспектральной информации и исследования различных сельскохозяйственных особенностей на местности.
3.2. Эмпирические соотношения
Линейная регрессия — широко используемый метод анализа гиперспектральных изображений и получения целевой информации (например, свойств сельскохозяйственных культур и почвы). Как спектральная отражательная способность, так и индексы растительности могут использоваться в качестве переменных-предикторов при установлении линейной зависимости. Например, используя спектральные полосы, Финн и др. [108] построили линейные регрессии между данными полевых измерений влажности почвы и спектральной отражательной способностью собранных гиперспектральных изображений и определили полосы, которые имеют более сильную корреляцию с влажностью почвы. В большем количестве исследований в регрессии использовались индексы растительности для лучшей производительности, поскольку некоторые индексы могут усиливать сигнал целевых объектов и минимизировать фоновый шум. Некоторые из предыдущих исследований показаны в Таблице 6.
В целом, линейная регрессия обычно использовалась для оценки широкого спектра свойств сельскохозяйственных культур или почвы. Ее легко установить, и большинство регрессий на основе индексов дали удовлетворительную точность. Однако с этим подходом связано несколько потенциальных проблем, таких как большое количество доступных индексов и неизвестность того, какой из них работает лучше, регрессия может быть очень чувствительна к размеру и качеству данных, а также проблема насыщения индексов [36,165]. Таким образом, крайне важно учитывать эти потенциальные проблемы и принимать соответствующие решения при установлении линейных регрессий с гиперспектральными данными. Например, рекомендуется выбирать соответствующие индексы растительности с целевыми переменными сельскохозяйственных культур или почвы. Исследователи оценили широкий спектр гиперспектральных индексов растительности для различных исследовательских целей. Хабудан и др. [166] исследовал и 11 гиперспектральных индексов растительности для оценки содержания хлорофилла в сельскохозяйственных культурах. Мейн и др. [167] исследовали 73 индекса растит ельности для оценки содержания хлорофилла в сельскохозяйственных культурах и видах деревьев саванны. Пэн и Гительсон [168] протестировали 10 мультиспектральных индексов и 4 гиперспектральных индекса для количественной оценки валовой первичной продуктивности сельскохозяйственных культур. Крофт и др. [169] проанализировали 47 гиперспектральных индексов для оценки содержания хлорофилла в листьях различных видов деревьев. Чжоу и др. [170] оценили восемь гиперспектральных индексов для оценки содержания азота в пшенице на уровне полога. Тонг и Хэ [165] оценили 21 мультиспектральный и 123 гиперспектральных индекса растительности для расчета содержания хлорофилла в траве как в масштабах листьев, так и полога. Юэ и др. [171] исследовали 54 гиперспектральных индекса растительности для оценки биомассы озимой пшеницы. Индексы работали по-разному в этих исследованиях; поэтому предлагается оценить наиболее эффективные из них в этих исследованиях и выбрать тот, который обеспечивает самую высокую точность.
3.3. Radiative Transfer Modelling
Моделирование переноса излучения — это физически обоснованный подход, который использует физические законы для моделирования взаимодействия электромагнитного излучения с растительностью (например, отражение, передача и поглощение) [180]. RTM моделируют спектры растительности (например, отражательную способность и пропускаемость листьев), используя биофизические и биохимические свойства растительности (например, содержание хлорофилла и воды) в прямом режиме, а также для инверсии этих переменных из спектральных измерений в обратном режиме [181]. PROSAIL — один из наиболее широко используемых RTM. Эта модель представляет собой интеграцию модели PROSPECT на уровне листьев и модели SAIL на уровне полога и способна моделировать отражательную способность полога, используя свойства листьев (например, содержание хлорофилла и воды), структурные параметры полога (например, LAI и угол наклона листьев) и отражательную способность почвы [18].
PROSAIL также использовался в сельскохозяйственных условиях для исследования свойств сельскохозяйственных культур и почвы. Например, Каса и Джонс [182] инвертировали PROSAIL и модель полога с трассировкой лучей с данными гиперспектрального отражения, измеренными спектрорадиометром, и данными гиперспектрального изображения, полученными с помощью спектрометра визуализации, соответственно, для оценки LAI полога и оценили факторы, влияющие на точность оценки (например, неоднородная поверхность, вызванная структурой ряда сельскохозяйственных культур). Рихтер и др. [98] использовали PROSAIL для оценки LAI, fCover, хлорофилла полога и содержания воды из гиперспектральных изображений и сравнили его производительность с другими методами (например, искусственной нейронной сетью). Рихтер и др. [183] применили PROSAIL для исследования аналогичных переменных растительности и проанализировали точность и эффективность этого метода. Ву и др. [184] исследовали чувствительность индексов растительности к содержанию хлорофилла в растительности, используя результаты моделирования модели PROSPECT, и предложили несколько хорошо работающих индексов. Лохерер и др. [74] попытались оценить LAI растительности, используя модель PROSAIL и гиперспектральные изображения из нескольких источников, и протестировали несколько методов (например, различные функции стоимости и типы методов усреднения), используемых для процесса инверсии. Ю и др. [37] оценили ряд переменных фенотипирования растительности (например, LAI и хлорофилл листьев), используя гиперспектральные изображения и PROSAIL, и исследовали чувствительность различных спектральных диапазонов к параметрам в модели PROSAIL.
По сравнению с моделями регрессии, обсуждавшимися в предыдущих разделах, модели RTM использовались в литературе реже для исследования сельскохозяйственных особенностей, в основном из-за их высокой сложности модели и вычислительной интенсивности. Например, в модели RTM необходимо учитывать широкий спектр параметров (например, хлорофилл, каротиноиды, содержание воды, индекс площади листьев, углы листьев, солнечные углы и отражательная способность почвы, а также другие параметры в модели PROSAIL), и пользователям необходимо использовать различные методы (например, функцию заслуг, таблицу поиска) для упрощения операций прямой и обратной модели. Кроме того, для достижения прогнозов целевых переменных растительности требуется гораздо больше вычислительного времени, чем в моделях регрессии. Однако также хорошо известно, что модели регрессии, как правило, привязаны к месту и времени и нелегко переносимы в другие географические регионы или в другое время на участке [166]. Напротив, RTM является более переносимым подходом, поскольку он устанавливается на основе физических законов и не требует обучающих данных для перестроения модели. Кроме того, RTM позволяет оценить ряд свойств растительности в одной модели, в то время как регрессионные модели обычно могут оценить только одну переменную [36,185].
3.4. Машинное обучение и глубокое обучение
Алгоритмы машинного обучения, включая регрессию опорных векторов (SVM) и RF, являются мощными инструментами для анализа гиперспектральной информации, поскольку они могут эффективно обрабатывать большое количество переменных (например, спектральную отражательную способность и индексы растительности) [186]. Машинное обучение широко использовалось в области дистанционного зондирования для оценки свойств наземных объектов или классификации различных наземных покровов [36,114,187]. Исследователи также использовали различные алгоритмы машинного обучения и гиперспектральные изображения для сельскохозяйственных приложений. SVM был широко используемым алгоритмом в предыдущих исследованиях для целей прогнозирования или классификации. Например, Хонкаваара и др. [123] оценили биомассу сельскохозяйственных культур с помощью SVM и гиперспектральных изображений, полученных с помощью БПЛА. Бостан и др. [51] использовали SVM для классификации различных типов сельскохозяйственных культур и достигли высокой точности классификации. Ран и др. [93] использовали классификаторы KNN и SVM для исследования методов обработки почвы на сельскохозяйственных полях и сравнили их производительность. RF — еще один широко используемый алгоритм для исследования сельскохозяйственных особенностей с помощью гиперспектральных изображений. Например, Гао и др. [188] успешно классифицировали сорняки и кукурузу с помощью RF и лабораторных гиперспектральных изображений. Используя наземные данные гиперспектрального отражения, полученные спектрорадиометром ASD, Зигманн и Джармер [189] оценили производительность RF, SVM и PLSR для оценки LAI сельскохозяйственных культур и подтвердили хорошую производительность RF. Аналогичным образом, используя гиперспектральное отражение, Адам и др. [190] попытались обнаружить болезнь кукурузы с помощью модели RF. В целом, модели машинного обучения, как правило, обладают надежной производительностью для исследования сельскохозяйственных особенностей с помощью гиперспектральных изображений.
Глубокое обучение является подмножеством машинного обучения и расширяет машинное обучение, добавляя больше «глубины» (т. е. иерархического представления набора данных) в модель [191,192]. Это популярный подход в последние годы для распознавания закономерностей на изображениях дистанционного зондирования и, таким образом, для исследования различных наземных объектов. Глубокое обучение обычно используется в области дистанционного зондирования для классификации изображений, такой как классификация земельного покрова [193,194,195] и идентификация наземных объектов (например, зданий) [196]. Глубокое обучение также применяется в точном земледелии для решения сложных задач. Существующие исследования, например, изучают оценку урожайности с использованием CNN и многоспектральных изображений вместе с климатическими данными [197], обнаружение болезней растений с использованием CNN и изображений, полученных со смартфона [198], классификацию сельскохозяйственных культур с использованием 3-D CNN и многовременных многоспектральных изображений [199] и классификацию сельскохозяйственного покрова с использованием глубокой рекуррентной нейронной сети и многовременных изображений SAR [200]. Камиларис и Пренафета-Болду [191] рассмотрели применение глубокого обучения в сельском хозяйстве и производстве продуктов питания, хотя не все исследования использовали изображения дистанционного зондирования. Сингх и др. [201] рассмотрели ряд методов глубокого обучения и их применение, в частности, в фенотипировании растений. До сих пор глубокое обучение не было хорошо изучено для обработки и анализа изображений дистанционного зондирования, особенно гиперспектральных изображений, для сельскохозяйственных приложений. Учитывая возможности глубокого обучения для изучения особенностей изображений и богатую информацию в гиперспектральных изображениях, интеграция этих двух технологий имеет широкий спектр сельскохозяйственных приложений (например, классификация сельскохозяйственных культур, мониторинг сорняков, обнаружение заболеваний сельскохозяйственных культур и оценка стресса растений). Дальнейшие исследования в этих областях необходимы.
Машинное обучение или глубокое обучение способно обрабатывать многоисточниковые и многотипные данные [202]. Например, помимо многотипных изображений дистанционного зондирования (например, оптических, тепловых, LiDAR и радарных), другие источники данных, такие как погода, орошение и историческая информация об урожайности, также могут быть включены в процесс моделирования для возможной лучшей оценки целевых сельскохозяйственных характеристик [203]. Хотя модели машинного обучения и глубокого обучения являются мощными, также важно помнить, что эти модели требуют большого количества и высококачественных обучающих выборок для достижения надежных результатов [202]. Недостаточные обучающие наборы данных или данные с проблемами (например, неполнота данных, шум и смещения) могут привести к нежелательным результатам модели.
Подводя итог, можно сказать, что различные аналитические методы (например, линейная регрессия, расширенная регрессия, машинное обучение и глубокое обучение, а также RTM) имеют разные уровни сложности, производительности и переносимости. Более подробные сравнения этих методов приведены в Таблице 7. В целом, линейная регрессия является самым простым в использовании методом, и ее производительность в целом приемлема, хотя на этот метод может сильно влиять выбор предикторных переменных и качество выборочных данных. Расширенная регрессия (например, PLSR) в основном работает лучше, чем линейная регрессия, поскольку она включает в себя несколько переменных в модели и менее чувствительна к шуму данных. RTM (например, PROSAIL) способен производить несколько продуктов данных (например, хлорофилл, вода и LAI) с достаточно высокой точностью. Одним из существенных преимуществ этого метода является его высокая переносимость. Однако этот метод имеет самую высокую сложность, поскольку требует широкого диапазона параметров и обширного программирования. С точки зрения машинного обучения многие алгоритмы, такие как RF и SVM, хорошо зарекомендовали себя и в основном хорошо себя зарекомендовали в предыдущих исследованиях. Для достижения оптимальной производительности этому методу необходимы некоторые корректировки программирования и модели. Глубокое обучение — относительно новый метод, который в последние годы становится все более популярным. Соответствующий дизайн и программирование модели имеют решающее значение для этого подхода. Он также требует значительного объема обучающих данных и вычислительных ресурсов для достижения хорошей производительности модели.
4. Гиперспектральные приложения в сельском хозяйстве
Гиперспектральная съемка используется в сельском хозяйстве для самых разных целей, включая оценку биохимических свойств сельскохозяйственных культур (например, хлорофилла, каротиноидов и содержания воды) и биофизических свойств (например, LAI, биомассы) для понимания физиологического состояния растительности и прогнозирования урожайности, оценки состояния питательных веществ сельскохозяйственных культур (например, дефицита азота), мониторинга заболеваний сельскохозяйственных культур и исследования свойств почвы (например, влажности почвы, органического вещества почвы и углерода почвы). Предыдущие исследования также обобщили некоторые из вышеупомянутых применений гиперспектрального дистанционного зондирования в точном земледелии [4,84]. Таким образом, в этом разделе мы больше сосредоточимся на недавних гиперспектральных исследованиях и обобщим эти исследования в соответствии с конкретными приложениями.
4.1. Оценка биохимических и биофизических свойств урожая
Одним из важных гиперспектральных приложений в сельском хозяйстве является мониторинг состояния урожая путем извлечения биохимических и биофизических свойств урожая [8,99]. Например, содержание хлорофилла в листьях является важным биохимическим свойством, влияющим на фотосинтетическую способность растений и контролирующим урожайность урожая [99]. В предыдущих исследованиях Оппельт и Маузер [105] собирали данные AVIS для извлечения содержания хлорофилла и азота на поле озимой пшеницы. Аналогичным образом Мохарана и Дутта [43] использовали данные Hyperion для оценки содержания этих двух биохимических компонентов на рисовом поле. С другой стороны, LAI является фундаментальным биофизическим параметром растительности и тесно связан с биомассой и урожайностью урожая [98]. В предыдущих исследованиях гиперспектральное дистанционное зондирование использовалось для оценки LAI различных культур, и некоторые примеры исследований показаны в Таблице 8.
В дополнение к вышеупомянутым биохимическим и биофизическим свойствам растительности, содержание воды в культуре является критическим параметром для выявления водного стресса. Рихтер и др. [98] попытались оценить содержание воды в кукурузе, сахарной свекле и озимой пшенице, используя данные бортового HyMap. Мохарана и Дутта [204] исследовали водный стресс на рисовом поле и его изменения, используя изображения Hyperion, и указали, что оцененное с помощью дистанционного зондирования содержание воды хорошо совпадает с данными полевых наблюдений. Иззо и др. [128] оценили состояние воды в коммерческом винограднике, используя гиперспектральные данные, полученные с помощью БПЛА, и определили длины волн, чувствительные к содержанию воды в пологе. Саху и др. [4] обсудили применение данных гиперспектрального дистанционного зондирования для оценки водных характеристик сельскохозяйственных культур и перечислили несколько индексов растительности для расчета содержания воды.
Из обзора литературы можно узнать, что многие предыдущие исследования были сосредоточены на оценке содержания хлорофилла в культуре, LAI и содержания воды с исполь зованием гиперспектральных изображений, в то время как другие важные свойства культур, такие как каротиноиды, которые чувствительны к стрессу растений, изучены меньше. Кроме того, на производство культур влияют все эти свойства растительности (например, хлорофилл, вода и LAI). Помимо изучения пространственных и временных изменений каждого свойства, также важно оценить взаимосвязи между этими свойствами и глубже понять, как они влияют на рост и производство культур.
Оценка биомассы культур и прогнозирование урожайности также являются важными приложениями дистанционного зондирования, поскольку они будут способствовать пониманию урожайности культур и внедрению соответствующих мер управления [126]. Юэ и др. [124] использовали гиперспектральные изображения на основе БПЛА для оценки надземной биомассы озимой пшеницы. Янг [205] и Мариотто и др. [15] использовали как мультиспектральные, так и гиперспектральные данные для оценки урожайности и обнаружил и, что модель на основе гиперспектральных изображений показала лучшие результаты. Кроме того, остатки урожая, оставленные на поле, являются критически важными материалами, защищающими почву от водной и ветровой эрозии и влияющими на биохимические процессы в почве. Предыдущие исследования, такие как Bannari et al. [106], Galloza and Crawford [47], Bannari et al. [46], использовали различные гиперспектральные изображения для оценки остатков урожая на сельскохозяйственных угодьях
Помимо оценки биомассы и остатков урожая, еще одной темой исследования является изучение биоэнергии (например, биогаза), которая может быть получена из биомассы урожая. Thomas et al. [100] попытались оценить количество биогаза, которое может быть получено на единицу биомассы, используя данные HyMap с воздуха, и достигли удовлетворительных результатов. В целом, гиперспектральные изображения внесли большой вклад в оценку биомассы сельскохозяйственных культур, урожайности и других связанных характеристик (например, биоэнергии, остатков урожая). Поскольку биомасса сельскохозяйственных культур и урожайность сильно зависят от сельскохозяйственных практик (например, полива и обработки удобрениями), включение этих данных практики вместе с гиперспектральными изображениями в модель может потенциально дать лучшие результаты. Необходимо больше исследований в этой области.
4.3. Классификация изображений для определения типов культур, стадий роста, сорняков/инвазивных видов и стрессов/болезней
Помимо количественной оценки свойств сельскохозяйственных культур, гиперспектральные изображения также использовались для целей классификации, таких как дифференциация типов сельскохозяйственных культур, определение стадий роста сельскохозяйственных культур, классификация сорняков или инвазивных видов и обнаружение заболеваний [218]. Примеры предыдущих исследований приведены в Таблице 10. Различные сельскохозяйственные угодья или типы сельскохозяйственных культур имеют разные спектральные характеристики; следовательно, гиперспектральные изображения могут внести большой вклад в классификацию этих сельскохозяйственных особенностей.
4.4. Получение влажности почвы, плодородия и других физических или химических свойств
Сельскохозяйственные свойства почвы, включая влажность почвы, органическое вещество почвы, засоленность почвы и шероховатость, являются важными факторами, влияющими на рост урожая и конечную продукцию [7]. Гиперспектральное дистанционное зондирование может внести большой вклад в изучение этих факторов. Например, оценка влажности почвы является одной из самых популярных тем исследований. Финн и др. [108] оценили влажность почвы на трех разных глубинах, используя гиперспектральные изображения с воздуха и линейную регрессию, и обсудили вклад и ограничения гиперспектрального дистанционного зондирования для исследований влажности почвы. Каса и др. [229] исследовали содержание воды, глины и песка в почве, используя слияние изображений CHRIS-PROBA и геофизических данных почвы. Шошани и др. [7] суммировали четыре основных подхода к оценке влажности почвы: (1) радиолокационные методы; (2) расчеты радиационного баланса и температуры поверхности; (3) отражательная спообность в видимом, ближнем ИК и коротковолновом ИК диапазонах; и (4) интегративные методы, использующие несколько спектральных диапазонов. Хотя влажность почвы можно оценить с помощью оптических данных дистанционного зондирования, на нее часто влияет растительный покров. Интеграция многотипных данных дистанционного зондирования, например, данных SAR и тепловых данных, может, возможно, дать более точные оценки.
SOC является критически важным компонентом плодородия почвы, который в значительной степени контролирует как рост, так и урожайность сельскохозяйственных культур. Гиперспектральные данные предоставляют тонкие спектральные детали, которые имеют решающее значение для оценки содержания SOC. В предыдущих исследованиях для исследования SOC использовались гиперспектральные изображения, собранные различными платформами ( таблица 13). В целом, гиперспектральные изображения имеют высокий потенциал для оценки органического вещества почвы и углерода. Однако, подобно оценке влажности почвы, исследование органического вещества почвы и углерода может сильно зависеть от растительного покрова. Поэтому сбор гиперспектральных изображений в невегетационные сезоны может быть решением.
Данные гиперспектрального дистанционного зондирования также использовались для оценки других характеристик почвы, как показано в Таблице 14. Из этих исследований можно сделать вывод, что гиперспектральные изображения можно использовать для изучения широкого спектра характеристик почвы. Различные характеристики почвы влияют на спектральные сигналы в разных диапазонах и с разной величиной, при этом некоторые из этих влияний могут спектрально перекрываться. Поэтому при исследовании определенной характеристики почвы крайне важно собрать подходящее количество образцов почвы, при этом другие характеристики почвы обычно контролируются.
Подводя итог, можно сказать, что гиперспектральная съемка успешно применялась в широком спектре сельскохозяйственных приложений, как рассмотрено выше и обобщено в Таблице 15. Также предлагаются направления будущих исследований.
5. Выводы и рекомендации
Гиперспектральная съемка имеет большой потенциал для применения в сельском хозяйстве, особенно в точном земледелии, благодаря обширной спектральной информации, чувствительной к различным биофизическим и биохимическим свойствам растений и почвы. Несколько платформ, включая спутники, самолеты, БПЛА и платформы ближнего действия, стали более широко доступны в последние годы для сбора гиперспектральных изображений с различным пространственным, временным и спектральным разрешением. Эти платформы также имеют различные сильные стороны и ограничения с точки зрения пространственного покрытия, продолжительности полета, гибкости, эксплуатационной сложности и стоимости. Эти факторы необходимо учитывать при выборе платформы(-й) съемки для конкретных исследовательских целей. Также необходимы дальнейшие технологические разработки для преодоления некоторых ограничений, таких как короткий срок службы батареи в операциях БПЛА и высокая стоимость гиперспектральных датчиков.
Различные аналитические методы, такие как линейная регрессия, расширенная регрессия, машинное обучение, глубокое обучение и RTM, были изучены в предыдущих исследованиях для анализа огромного количества информации в гиперспектральных изображениях для исследования различных сельскохозяйственных объектов. Предыдущие исследования в основном использовали регрессионный подход, в то время как более физически обоснованные методы, такие как RTM, были изучены меньше. Глубокое обучение и эффективная аналитика больших данных являются мощными инструментами для распознавания закономерностей в данных дистанционного зондирования. Вместе с гиперспектральными изображениями модели глубокого обучения имеют высокий потенциал для поддержки мониторинга широкого спектра сельскохозяйственных объектов. Различные аналитические методы имеют разные преимущества и недостатки, и поэтому важно сравнивать эти методы для конкретных исследований (например, требования к точности и вычислительной эффективности) и выбирать оптимальный подход. Кроме того, спектральная информация изображения обычно использовалась в качестве переменных для задач прогнозирования или классификации, в то время как другая информация, такая как текстура, была изучена меньше. Кроме того, некоторые другие источники данных, такие как погода, записи об орошении и историческая информация об урожайности, также могут использоваться в некоторых аналитических методах (например, машинное обучение и глубокое обучение) для лучшего мониторинга характеристик сельскохозяйственных культур. Также необходимы дополнительные исследования в этих областях.
Гиперспектральная визуализация успешно применялась в широком спектре сельскохозяйственных приложений, включая оценку биохимических и биофизических свойств сельскохозяйственных культур; оценку питательных веществ и стрессового статуса сельскохозяйственных культур; классификацию или обнаружение типов сельскохозяйственных культур, сорняков и болезней; и исследование характеристик почвы. Предыдущие исследования были сосредоточены на обсуждении одного или двух из многих факторов, влияющ их на производительность и продуктивность сельскохозяйственных культур, и, таким образом, не могут всесторонне оценить состояние сельскохозяйственных культур и факторы, ограничивающие рост. Важно интегрировать эти факторы, чтобы достичь лучшего понимания их взаимосвязей для оптимального производства сельскохозяйственных культур и защиты окружающей среды. Кроме того, предыдущие исследования с использованием гиперспектральной визуализации были в основном направлены на изучение роста сельскохозяйственных культур с целью повышения урожайности сельскохозяйственных культур, в то время как меньше исследований было сосредоточено на понимании экосистемной стороны сельскохозяйственного производства (например, экосистемных услуг и биоразнообразия). Необходимы дальнейшие исследования в этих областях.
Вклад авторов
Концептуализация, J.S., J.L., Y.H., B.L. и P.D.D.; методология, B.L., P.D.D. и Y.H.; расследование, B.L.; написание — подготовка первоначального черновика, B.L.; написание — рецензирование и редактирование, P.D.D., J.S., J.L. и Y.H.; администрирование проекта, J.S, J.L. и Y.H.; получение финансирования, Y.H. Все авторы прочитали и согласились с опубликованной версией рукописи.
Финансирование
Данная работа финансировалась Канадским советом по естественным наукам и инженерным исследованиям (NSERC) в рамках исследовательского гранта RGPIN-386183, предоставленного профессору Юйхонгу Хэ.
Конфликты интересов
Авторы заявляют об отсутствии конфликта интересов.
Сокращения
|
ALI |
Advanced Land Imager |
|
APEX |
Airborne Prism Experiment |
|
AVIS |
Airborne Visible Near-Infrared Imaging Spectrometer |
|
AVIS |
Airborne Visible Near-Infrared Imaging Spectrometer |
|
AVIRIS |
Airborne Visible/Infrared Imaging Spectrometer |
|
ANN |
Artificial Neural Networks |
|
CAI |
Cellulose Absorption Index |
|
CAI |
Chlorophyll Absorption Integral |
|
CARI |
Chlorophyll Absorption Ratio Index |
|
CASI |
Compact Airborne Spectrographic Imager |
|
CHRIS |
Compact High Resolution Imaging Spectrometer |
|
CNN |
Convolutional Neural Network |
|
DEM |
Digital Elevation Model |
|
DESIS |
Dlr Earth Sensing Imaging Spectrometer |
|
DCNI |
Double-Peak Canopy Nitrogen Index |
|
EnMAP |
Environmental Mapping And Analysis Program |
|
FAPAR |
Fraction Of Absorbed Photosynthetically Active Radiation |
|
fCover |
Fraction Of Vegetation Cover |
|
GCPs |
Ground Control Points |
|
HSI |
Hyper Spectral Imaging |
|
HySI |
Hyperspectral Imager |
|
HICO |
Hyperspectral Imager For The Coastal Ocean |
|
HISUI |
Hyperspectral Imager Suite |
|
HyspIRI |
Hyperspectral Infrared Imager |
|
HyMap |
Hyperspectral Mapper |
|
h NDVI |
Hyperspectral Normalized Difference Vegetation Index |
|
PRISMA |
Hyperspectral Precursor And Application Mission |
|
IMU |
Inertial Measurement Unit |
|
LAI |
Leaf Area Index |
|
MTCI |
Meris Terrestrial Chlorophyll Index |
|
MNF |
Minimum Noise Fraction |
|
MCARI/MTVI2 |
Modified Chlorophyll Absorption Ratio Index/Modified Triangular Vegetation Index 2 |
|
MSR |
Modified Simple Ratio Index |
|
MSAVI |
Modified Soil Adjusted Vegetation Index |
|
MTVI2 |
Modified Triangular Vegetation Index |
|
MIVIS |
Multispectral Infrared Visible Imaging Spectrometer |
|
MSI |
Multispectral Instrument |
|
MLR |
Multi-Variable Regression |
|
NDRE |
Normalized Difference Red Edge |
|
NDTI |
Normalized Difference Tillage Index |
|
OLI |
Operational Land Imager |
|
OSAVI |
Optimized Soil-Adjusted Vegetation Index |
|
PLSR |
Partial Least Square Regression |
|
PRI |
Photochemical Reflectance Index |
|
PRESS |
Predicted Residual Error Sum Of Squares |
|
PCA |
Principal Component Analysis |
|
PHI |
Pushbroom Hyperspectral Imager |
|
RTM |
Radiative Transfer Modelling |
|
RF |
Random Forest |
|
REP |
Red Edge Position |
|
SWIR |
Shortwave Infrared |
|
SR |
Simple Ratio |
|
SVD |
Singular Value Decomposition |
|
SOC |
Soil Organic Carbon |
|
SHALOM |
Spaceborne Hyperspectral Applicative Land And Ocean Mission |
|
SFOC |
Special Flight Operations Certificate |
|
SVM |
Support Vector Machine Regression |
|
TCARI |
Transformed Chlorophyll Absorption In Reflectance Index |
|
TCI |
Triangular Chlorophyll Index |
|
TVI |
Triangular Vegetation Index |
|
UMD |
Uniform Feature Design |
|
UAV |
Unmanned Aerial Vehicle |
- Категория: Блог
- Просмотров: 289
Цена оказывает существенное влияние на поведение потребителей, и с точки зрения экономии денег люди всегда хотят покупать продукты, которые имеют хорошее соотношение цены и качества. Так стоит ли гиперспектральная камера дорого? Как купить гиперспектральную камеру с хорошим соотношением цены и качества? В этой статье представлен краткий анализ.
Дорого ли стоят гиперспектральные камеры?
Гиперспектральные камеры сильно различаются по цене в зависимости от таких факторов, как марка, модель, производительность и использование. В целом, по сравнению с обычными камерами, гиперспектральные камеры дороже, обычно от нескольких тысяч до десятков тысяч долларов, но также доступны не которые гиперспектральные камеры низкого и среднего диапазона.
Основная причина высокой цены гиперспектральных камер заключается в том, что их производственный процесс и технология выше, что требует более высокой точности и стабильности. Гиперспектральные камеры обычно должны быть оснащены более совершенными объективами и датчиками для захвата данных изображения с высоким разрешением в сверхшироком спектральном диапазоне. Кроме того, для обеспечения точности и надежности камер требуются процедуры проверки качества, такие как калибровка и тестирование. Все эти факторы увеличивают стоимость производства и продажную цену.
Как снизить цены на гиперспектральные камеры
1. Выберите правильную модель: выберите модель, которая подходит для вашего сценария применения, и не нужно покупать слишком дорогие продукты.
2. Дождитесь рекламных акций: гиперспектральные камеры имеют узкий диапазон применения, поэтому вы можете дождаться рекламных акций продавцов
соответствующим образом.
3. Покупка подержанных или старых моделей: вы можете выбрать покупку подержанной или старой гиперспектральной камеры, по сравнению с новой
моделью цена будет ниже.
Выбирать и приобретать гиперспектральные камеры нужно внимательно и полностью учитывать множество факторов, включая бренд, производительность
и функции и т. д., чтобы выбрать подходящий для вас продукт.
Как выбрать гиперспектральную камеру
1. Выбор на основе сценариев применения: гиперспектральные камеры обычно используются в таких областях, как сельское хозяйство, окружающая среда и спутниковая съемка. В зависимости от конкретной сцены следует определить производительность и функции гиперспектральной камеры. 2.
2. выбор по бюджету: цена гиперспектральной камеры высока, в зависимости от диапазона бюджета следует определить, какой класс гиперспектральной камеры можно приобрести.
3. Выбор надежного бренда: на рынке представлены различные бренды гиперспектральных камер, выберите бренд с высокой видимостью и гарантированным качеством.
Преимущества гиперспектральных камер
1. Высокое разрешение: гиперспектральные камеры могут захватывать более широкий спектральный диапазон, чем обычные камеры, и имеют более высокое пространственное разрешение, что может обеспечить более богатую информацию об изображении.
2. Многоспектральная визуализация: гиперспектральные камеры могут создавать изображения в нескольких спектральных диапазонах, что помогает различать различные типы света, тем самым улучшая анализ и идентификацию изображений.
3. Точный количественный анализ: поскольку гиперспектральные камеры могут предоставлять больше спектральной информации, они могут более точно количественно анализировать состав и свойства объектов.
4. Широкое применение: гиперспектральные камеры имеют широкий спектр применения в сельском хозяйстве, мониторинге окружающей среды, геологи ческой разведке, медицинской диагностике и других областях.
- Категория: Блог
- Просмотров: 349
Спектральный, мультиспектральный, гиперспектральный, не можете заметить разницу?

Спектральный анализ как важное средство естественнонаучного анализа, спектральная технология часто используется для обнаружения физической структуры объектов, химического состава и других показателей. Спектрометрия изображений, с другой стороны, объединяет спектральную технологию и технологию визуализации, объединяя спектральное разрешение и графическое разрешение, что приводит к многогранному спектральному анализу в пространственном измерении, который теперь известен как технология мультиспектральной визуализации и гиперспектральной визуализации.
В чем разница между спектральным, мультиспектральным и гиперспектральным?
Спектр
Спектр — это монохроматический свет, разделенный дисперсией после дисперсионной системы (например, призмы, решетки), через систему формирования изображений, проецируемый на детектор, чтобы стать размером длины волны (или частоты) последовательного расположения рисунка, который известен как оптический спектр. Спектрометр Ocean Optics основан на этом принципе проектирования и производства.
В зависимости от длины волны световые волны имеют разные названия: длины волн в диапазоне 380 и 780 нм называются видимым светом, волны короче 380 нм называются ультрафиолетовым светом, а волны длиннее 780 нм называются инфракрасным светом (инфракрасный свет также делится на ближний инфракрасный, средний инфракрасный, дальний инфракрасный и т. д.).
Мультиспектральный
Мультиспектральная технология относится к одновременному получению нескольких оптических спектральных диапазонов (обычно больше или равно 3), а также в видимом свете на основе инфракрасного света и ультрафиолетового света для расширения направления технологии спектрального обнаружения. Обычный метод реализации заключается в использовании различных фильтров или светоделителей и различных комбинаций фотопленки, чтобы в то же время, соответственно, получать одну и ту же цель в диапазоне различных узких спектральных диапазонов излучаемых или отражаемых световых сигналов, чтобы получить цель в нескольких различных спектральных диапазонах фотографии. Наиболее распространенными мультиспектральными фотографиями являются те, которые сделаны цветными камерами, как показано ниже, которые содержат информацию в трех оптических спектральных диапазонах, красном (1), зеленом (2) и синем (3), со спектральной точки зрения. Если к камере или детектору добавить больше диапазонов, таких как диапазоны (4) и (5), можно получить мультиспектральный снимок с несколькими диапазонами.
Мультиспектральная технология в сочетании с оборудованием для формирования изображений позволяет представлять мультиспектральную информацию в виде изображения.
Конечно, также возможно использовать только детектор для получения спектральной информации одной пространственной точки. Pixelteq, бренд Ocean Optics, с его уникальной технологией фильтрации чипов, может реализовать получение 8 каналов спектральной информации на чипе размером 9*9 см, что особенно подходит для приложений с чрезвычайно высокими требованиями к пространству и стоимости.
Гипеспектральный
Гиперспектроскопия — это тонкая технология, которая позволяет захватывать и анализировать спектры по точкам в пространственной области благодаря уникальным спектральным «особенностям», которые можно обнаружить в различных пространственных точках одного объекта, и, следовательно, может обнаруживать вещества, которые невозможно различить визуально.
Пример гиперспектрального изображения: изображения состоят из более узких полос (10-20 нм). Гиперспектральные изображения могут иметь сотни или тысячи полос. После того, как объект взаимодействует со светом от источника света и принимается невизуальным спектральным анализирующим устройством (например, спектрометром), устройство может точно реагировать на различия в интенсивности распределения принятого светового сигнала по спектральным полосам, также известным как спектральная информация. При использовании гиперспектрального оборудования, с точки зрения характеристик изображения, вы можете понять спектральную информацию каждой позиции образца, с точки зрения спектральных характеристик, вы можете понять распределение позиции сигнала в определенной спектральной полосе, то есть гиперспектральное оборудование может получить более богатую подробную информацию. Например: человеческий глаз может воспринимать только три спектральные полосы в сигнале световой энергии объекта: красный, зеленый и синий. То есть, мы часто называем это тремя основными цветами, но на самом деле мы можем видеть комбинацию этих трех цветов, создаваемую оранжевым, фиолетовым, лаймово-зеленым и так далее более тонкими цветами. Однако мы не можем различить разницу между чистым желтым и смесью красного и зеленого, которая также известна как «изохроматическая». Но гиперспектральная визуализация может легко различить разницу.
Выше два желтых цвета, один «сплошной цвет», а другой — смесь красного и зеленого, могут быть визуально неразличимы, но из-за их спектральных различий их можно различить с помощью спектроскопического оборудования. В наших экспериментах данные, полученные с помощью спектрометра, представляют собой среднее значение света, испускаемого всеми молекулами, взаимодействующими с источником падающего света во всем обнаруженном диапазоне, тогда как с помощью многоспектрального устройства можно получить информацию об образцах в нескольких определенных полосах в различных точках в пределах обнаруженного диапазона. В результате ни одно из этих устройств не может предоставить очень точную информацию об образце в одной области.
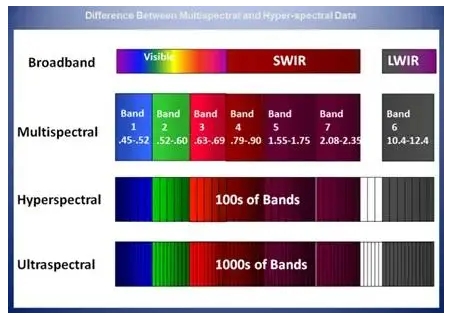
Гиперспектральный формирователь изображений (HSI) можно сравнить с сотнями или тысячами одноточечных спектрометров, выстроенных в ряд и одновременно фокусирующихся на области, при этом каждый спектрометр работает независимо и получает спектральную информацию о своем собственном местоположении. Выходные данные HSI представляют собой изображение или видеопоток, в котором каждый пиксель имеет свой собственный спектр, а каждый спектр содержит сотни спектральных полос. Эта «полноспектральная» возможность гиперспектральной визуализации позволяет видеть спектральные сигналы в каждом различимом пространственном месте сцены, т. е. получается больше размерной информации. Таким образом, гиперспектральную визуализацию можно использовать в различных приложениях, включая идентификацию произведений искусства, здоровье сельскохозяйственных культур, картографирование береговой линии, лесное хозяйство, разведку полезных ископаемых, городскую и промышленную инфраструктуру, качество продукции на производственных линиях, мониторинг окружающей среды и многое другое.
Методы гиперспектрального сканирования и результаты визуализации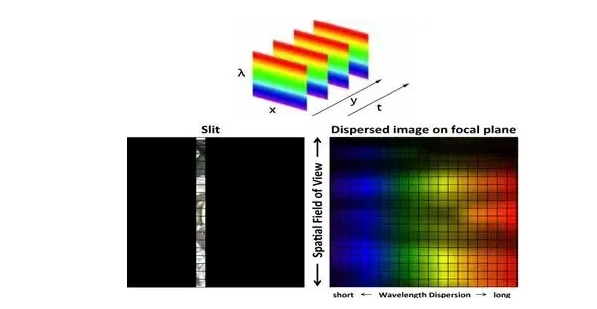
Разница между гиперспектральным и мультиспектральным
Очень часто спектр отражательной способности материала может быть очень сложным по отношению к длине волны, и другие мельчайшие особенности могут быть неразличимы с помощью более грубых методов многоспектральной визуализации.
Вещества, которые были неотличимы от тех, которые были идентифицированы с помощью мультиспектральной визуализации (слева) на рисунке выше, были различены с помощью гиперспектральной визуализации (справа). Причина этого в том, что поскольку гиперспектральная визуализация имеет больше спектральных полос, более сложные признаки отпечатков пальцев могут быть точно получены с более высоким спектральным разрешением.
Типичные применения
Гиперспектральные устройства могут обнаруживать определенные краски или красители в инфракрасном диапазоне, которые не видны человеческому глазу. Аналогично, системы HSI в диапазоне 60 или 300 могут предоставить более богатую спектральную информацию об отражательной способности материала, чем многоспектральная система, что позволяет более точно характеризовать материал. На изображении ниже показано изображение и спектральная информация, полученные с образца свежей ткани животного, помещенного на конвейерную ленту в лаборатории с использованием гиперспектрального формирователя изображений:
Спектрограммы различных областей: (а) маркированные области чистого жира, мраморности и чисто постных частей на образцах тканей; (б) Спектрограммы, маркированные в различных областях диаграммы (а).
Кроме того, мы можем предоставить интуитивно понятные программы для анализа изображений, классификации и визуализации различных веществ с уникальными спектральными характеристиками. Независимо от того, получены ли эти данные с воздуха, с земли или в лаборатории, вы можете увидеть на экране компьютера детали, которые могут быть неразличимы глазом.
- Категория: Блог
- Просмотров: 310
Гиперспектральное дистанционное зондирование является современным передовым этапом технологии дистанционного зондирования, использование множества очень узких диапазонов электромагнитных волн от интересующего объекта для получения соответствующих данных, содержит богатство пространственной, радиометрической и спектральной тройной информации, разработка которой является революцией в дистанционном зондировании, но также вызвала фундаментальные изменения в методах обработки данных и анализа информации, так что оригинал в широкополосном дистанционном зондировании в необнаруживаемом материале, в гиперспектральном дистанционном зондировании может быть обнаружен. Так что же такое гиперспектральный? Он начинается с видимого диапазона солнца.
Почему мир такой красочный?
Будь то юг неба или смена сезонов, почему мы чувствуем, что мир такой красочный? Радуга, которая проясняется после дождливого дня, может помочь нам найти ответ. Солнечный свет на самом деле является смешанным светом, через рассеивание капель воды в воздухе разлагается на красочный монохроматический свет, составляющий все цвета, которые могут воспринимать наши глаза, эта часть света определяется как видимые длины волн солнца.
В дополнение к этому солнечный свет также содержит свет в ультрафиолетовых и инфракрасных длинах волн. Однако тот же цвет света для наших человеческих глаз также является сложным цветом света, содержащим тысячи полос, что находится далеко за пределами предела человеческого глаза, чтобы различать. В то же время разные объекты состоят из разных элементов и их соединений, структура материала также различна, что приводит к тому, что длина волны отраженного или рассеянного света на поверхности объекта также показывает специфику; разные объекты в разных состояниях с разными длинами волн света отражают или рассеивают разную способность, но также делают объект имеющим разный цвет или спектральные характеристики, как и «отпечатки пальцев». Подобно информации «отпечатков пальцев», она может различать особенности и состав атмосферы тонким способом. Такие уникальные спектральные характеристики веществ составляют основу для идентификации и анализа характеристик различных объектов в науке дистанционного зондирования.

Для точного получения световой информации в различных диапазонах длин волн оптические дистанционные датчики спутников последовательно приняли технологию многоспектральной визуализации и технологию гиперспектральной визуализации, в которой свет, отраженный или рассеянный от объектов, разделяется на определенные диапазоны длин волн света с помощью фильтров, призм, решеток и других устройств для разделения света, а цели идентифицируются, а дистанционное зондирование количественно оценивается на основе информации о спектральных характеристиках, полученной с земли или от атмосферного отражения или рассеивания.
Первоначально дистанционное зондирование земли использовало систему технологии многоспектральной визуализации, часто всего с несколькими каналами, каждый канал содержит оптическую информацию с длинами волн шириной в десятки нанометров и может реализовать спектральную способность обнаружения как в инфракрасном, так и в ультрафиолетовом направлениях. Однако для схожих объектов или объектов в разных состояниях характерные пики их спектров отражения обычно схожи, как показано на рисунке ниже, характерные пики четырех видов деревьев лишь немного отличаются на 960 нанометрах, и для того, чтобы различать категории разных видов деревьев, требуется спектральное разрешение менее десяти нанометров; например, для классификации и идентификации горных пород, появления вредителей и болезней сельскохозяйственных культур, восстановления почвы для сельского хозяйства и вспышек флоры или цианобактерий, например, для классификации и идентификации горных пород, вредителей и болезней сельскохозяйственных культур, восстановления и обработки почвы, цветения воды или вспышек цианобактерий, загрязнения воздуха и других проблем, которые отражаются в спектрах только при изменении в несколько нанометров, традиционные средства многоспектрального обнаружения перегружены, и процент успешной идентификации невысок.
 С началом разработки гиперспектральной технологии дистанционного зондирования в 1970-х годах область оптического дистанционного зондирования претерпела революционные изменения и постепенно сформировала популярную область передовых технологий. Гиперспектральная технология дистанционного зондирования - это технология, основанная на очень многих узкополосных данных изображения, которая объединяет технологию визуализации со спектральной технологией для обнаружения двумерного геометрического пространства и одномерной спектральной информации цели, а также для получения непрерывных узкополосных данных изображения с высоким спектральным разрешением. Технология гиперспектральной визуализации быстро развивается, и к распространенным из них относятся решеточная спектроскопия, акустооптическая спектроскопия с перестраиваемым фильтром, призменная спектроскопия и покрытие чипа.
С началом разработки гиперспектральной технологии дистанционного зондирования в 1970-х годах область оптического дистанционного зондирования претерпела революционные изменения и постепенно сформировала популярную область передовых технологий. Гиперспектральная технология дистанционного зондирования - это технология, основанная на очень многих узкополосных данных изображения, которая объединяет технологию визуализации со спектральной технологией для обнаружения двумерного геометрического пространства и одномерной спектральной информации цели, а также для получения непрерывных узкополосных данных изображения с высоким спектральным разрешением. Технология гиперспектральной визуализации быстро развивается, и к распространенным из них относятся решеточная спектроскопия, акустооптическая спектроскопия с перестраиваемым фильтром, призменная спектроскопия и покрытие чипа.
Каковы технические характеристики гиперспектрального дистанционного зондирования?
Гиперспектральная (гиперспектральная камера) одноканальная полоса пропускания узкая, ее спектральное разрешение достигает порядка нанометра (нм) (обычно менее 10 нм), количество спектральных каналов достигает десятков или даже сотен, что позволяет получать непрерывные спектральные данные по отражению/рассеиванию материала, которые могут быть реализованы в диапазоне видимого, ближнего инфракрасного, среднего инфракрасного и теплового инфракрасного диапазонов длин волн, а также получать гиперспектральные данные.
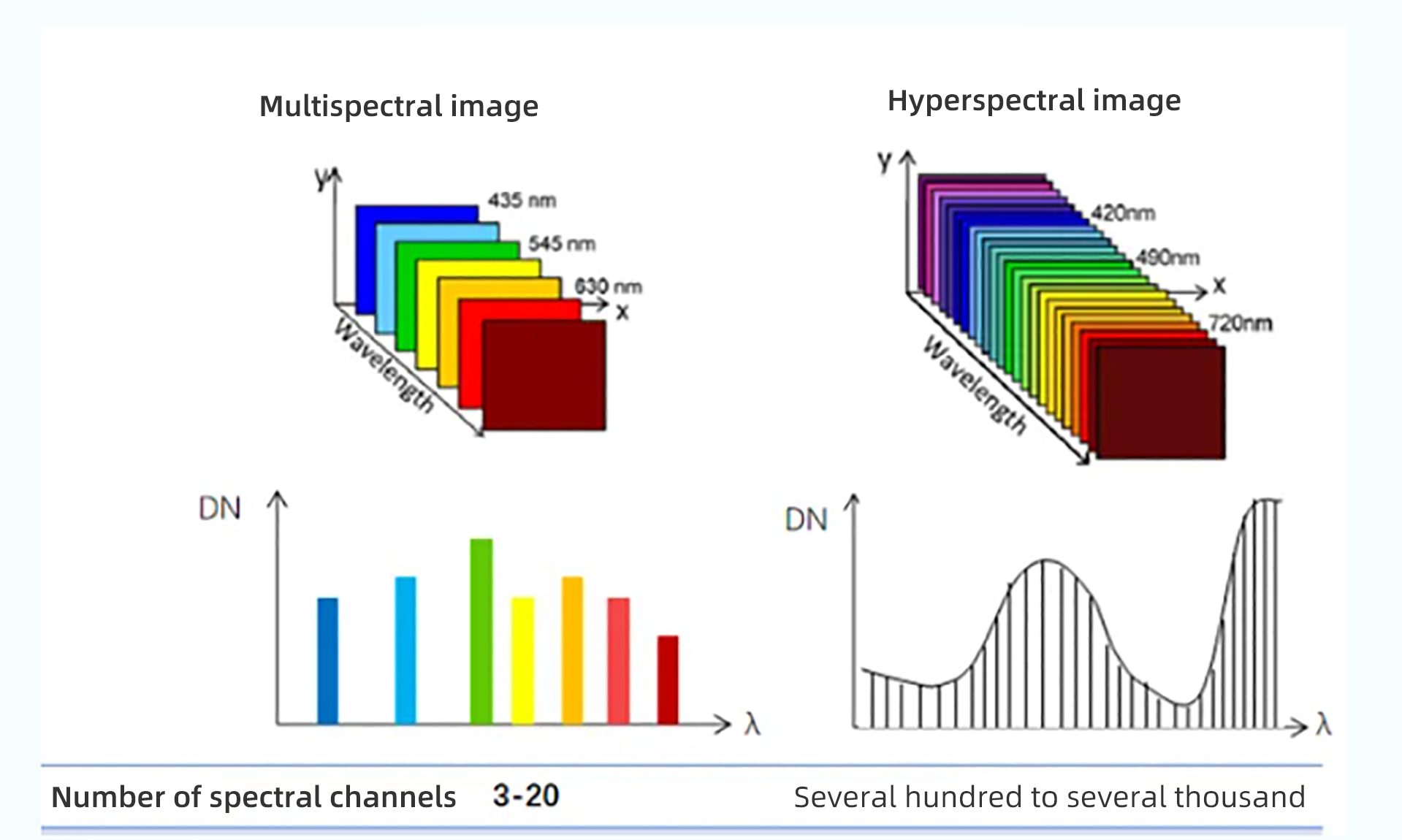
В отличие от традиционной технологии многоспектрального дистанционного зондирования, которая различает цели на основе цветовых различий, технология гиперспектрального дистанционного зондирования может достигать дискретной выборки в спектральном пространстве, и цели, которые можно различить, как правило, те, которые имеют очевидные различия в спектральном пространстве, такие как водоемы, растительность и голая земля. Гиперспектральное дистанционное зондирование представляет собой технологию получения многомерной информации, которая объединяет технологию визуализации и спектральную технологию, которая может одновременно получать двумерную пространственную информацию о цели и трехмерную спектральную информацию, и анализировать информацию о составе материала через морфологию спектральных кривых для идентификации цели, а также целевых особенностей, которые могут различать различные категории одного и того же типа особенностей. В соответствии с различными сценариями применения, селективность спектра становится гибкой и разнообразной, что улучшает способность различать и идентифицировать признаки, эффективно различает различные категории, принадлежащие к одному и тому же виду признаков, реализует «различные спектры для одного и того же вида признаков» и «различные признаки для одного и того же вида спектра» и уменьшает явление спектрально-пространственной путаницы признаков, такое как явление разных видов деревьев. Это может уменьшить явление спектрально-пространственной путаницы признаков, такое как идентификация разных видов деревьев и разных минералов; в то же время гиперспектральные данные могут использоваться для извлечения биофизических и химических параметров и биохимического анализа хлорофилла а, лигнина и целлюлозы растительности.
Развитие технологии гиперспектрального дистанционного зондирования превращает дистанционное зондирование из качественного анализа в количественное или полуколичественное преобразование. Основное применение традиционной технологии дистанционного зондирования с получением изображений основано на качественном анализе. Часть результатов количественного анализа точности результатов не идеальна, что, очевидно, связано со спектральным и пространственным разрешением датчика изображения, помехами атмосферного и почвенного фона и другими ограничениями. Дистанционное зондирование с получением изображений с гиперспектральным разрешением впервые преодолевает ограничение спектрального разрешения, что в значительной степени подавляет влияние других мешающих факторов в спектральном пространстве, что очень полезно для повышения точности результатов количественного анализа.
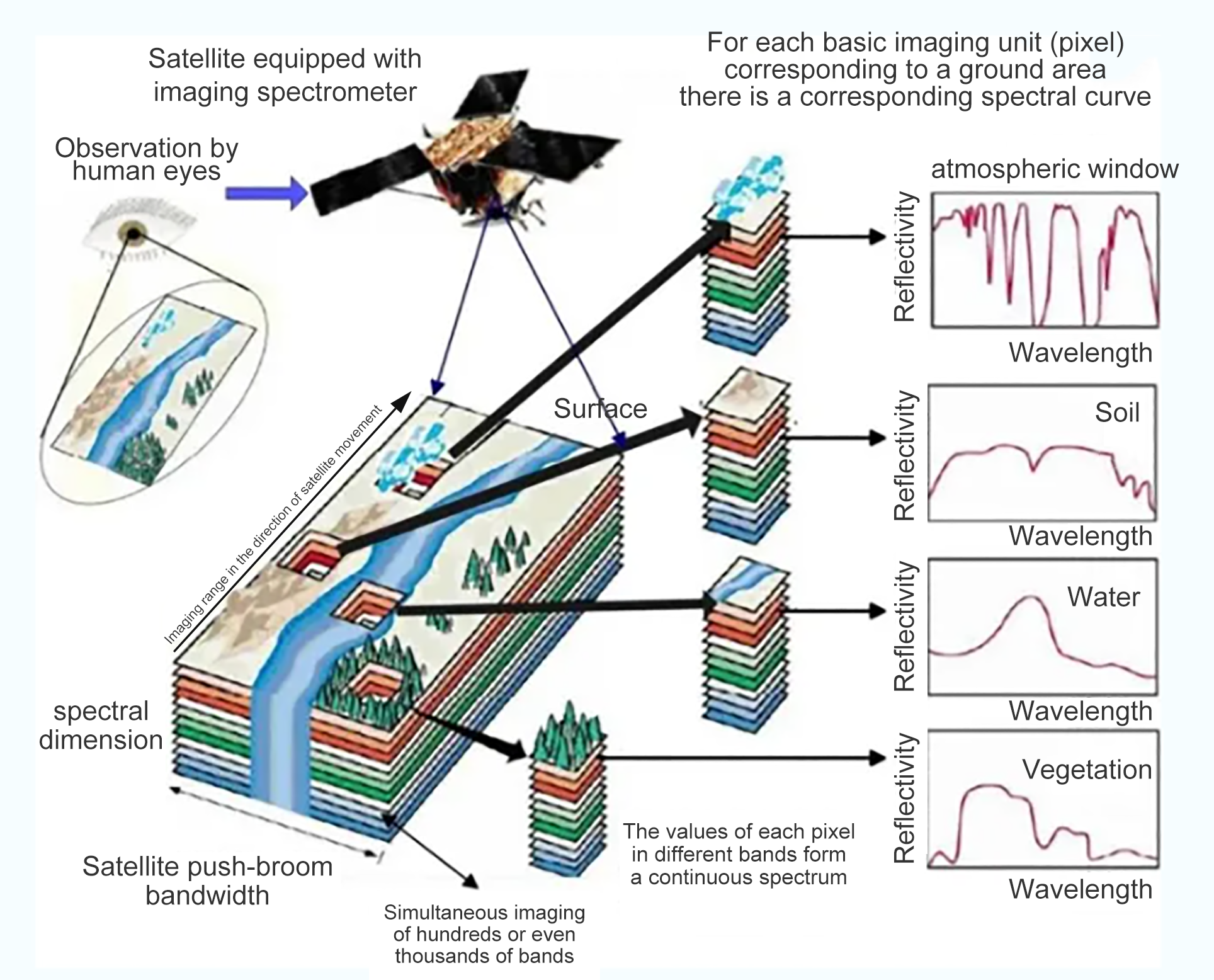
Что такое гиперспектральные приложения?
По сравнению с изображениями высокого разрешения и многоспектральными изображениями гиперспектральные изображения имеют высокое спектральное разрешение и множество полос, которые могут получать почти непрерывные спектральные кривые признаков, и конкретные полосы могут быть выбраны или извлечены в соответствии с необходимостью выделения целевых признаков; квантованные данные непрерывной спектральной кривой обеспечивают условия для введения классификации изображений в модель спектрального механизма признаков, которая содержит богатую радиометрическую, пространственную и спектральную информацию и является синтезом разнообразной информации. Она содержит богатую радиометрическую, пространственную и спектральную информацию и является всеобъемлющим носителем различной информации. Гиперспектральные изображения широко используются в областях геоморфологического картирования, разведки ресурсов, сельскохозяйственного дистанционного зондирования, экологического дистанционного зондирования, мониторинга лесного хозяйства, дистанционного зондирования почв, дистанционного зондирования цвета воды и атмосферной науки.
1. Дистанционное зондирование для классификации объектов
На рисунке показаны гиперспектральные данные прибрежной зоны Дубая, полученные с помощью гиперспектральных дистанционных датчиков, которые могут точно распознавать информацию об основных категориях объектов, таких как водоемы, здания, дороги, голая почва и т. д., посредством классификации объектов, и могут быть подразделены на 3–5 эффективных подкатегорий в каждой основной категории, а также могут распознавать информацию о судах в отдаленном море.
2. Разведка рудных месторождений
Технология гиперспектрального дистанционного зондирования может дать толчок геологической разведке. На основе анализа спектральных кривых полученных пород можно узнать типы распределения минералов и площадь места. На рисунке показано, что данные гиперспектрального дистанционного зондирования эффективно извлекли два вида минеральной информации о сериците и хлорите в районе Дулан провинции Цинхай, а после преобразования MNF они улучшают распознавание литологической и тектонической геологической информации.

3. Дистанционное зондирование водной среды
Из-за определенного сходства между спектрами водных растений и цветения воды и спектрами растительности, для обычно используемых многоспектральных данных дистанционного зондирования сложно точно идентифицировать цветение воды и водные растения, и только гиперспектральные данные дистанционного зондирования могут улавливать подробные спектральные различия между сложными и изменчивыми цветениями воды, водными растениями и водоемом, чтобы точно идентифицировать цветение воды и водные растения. На рисунке показана экологическая карта регионального водоема Юньнань Дяньчи, полученная с помощью спутникового гиперспектрального дистанционного датчика, который может четко определять окрашенное растворенное органическое вещество (CDOM), хлорофилл a (Chl-a) и концентрацию взвешенных твердых частиц (TSM) в водоеме. Между тем, параметры качества воды малых рек и озер, отличных от Дяньчи, также четко распознаются на изображении.
4. Дистанционное зондирование атмосферы
Дистанционный мониторинг выбросов метана из точечных источников проводился в Ливии и США с использованием гиперспектральных данных дистанционного зондирования с оптимизированными алгоритмами инверсии концентрации метана в столбе. На рисунке (a) показаны результаты мониторинга утечки метана из нефтяной скважины Dor Marada в Ливии, а на рисунке (b) показаны результаты мониторинга утечки метана из нефтяной скважины DCP Midstream в Пермском бассейне США, которая может точно контролировать чистый шлейф выбросов метана в регионе.

- Категория: Блог
- Просмотров: 306
Аннотация: Технология гиперспектральных изображений, как передовая технология дистанционного зондирования, достигла значительного развития и широкого применения во многих областях в последние годы. В этой статье подробно излагаются принципы и характеристики гиперспектральных изображений, подробно обсуждаются их применение в сельском хозяйстве, мониторинге окружающей среды, геологоразведке и других областях, а также рассматриваются будущие тенденции развития.
1. Введение
С непрерывным развитием науки и техники технология гиперспектральных изображений играет все более важную роль во многих областях благодаря своим уникальным преимуществам. Гиперспектральные изображения могут не только предоставлять богатую пространственную информацию, но и получать точную спектральную информацию, обеспечивая надежную поддержку для идентификации целей, классификации и количественного анализа.
2. Принципы и характеристики гиперспектральных изображений
(1) Принцип
Гиперспектральные изображения состоят из серии непрерывных узкополосных изображений, каждая полоса соответствует разному диапазону длин волн. Измеряя отражение, излучение и другие характеристики целевого объекта на разных длинах волн, можно получить кривую спектральной характеристики цели. Эти кривые спектральной характеристики содержат физическую, химическую и другую характерную информацию о цели и могут использоваться для идентификации и классификации цели.
(2) Особенности
Высокое спектральное разрешение: позволяет различать небольшие спектральные различия и предоставлять более богатую спектральную информацию.
Многополосная информация: содержит десятки или даже сотни полос, которые могут всесторонне отражать характеристики цели.
Сочетание пространственной информации и спектральной информации: позволяет не только получить пространственное распределение цели, но и понять ее спектральные характеристики.
Бесконтактное измерение: нет необходимости контактировать с целевым объектом, можно осуществлять мониторинг на большом расстоянии и большой площади.
3. Применение гиперспектральных изображений в различных областях
(1) Сельскохозяйственное поле
Мониторинг посевов: он может контролировать состояние роста посевов, вредителей и болезней и т. д., а также оказывать поддержку принятию решений для точного земледелия. Например, анализируя спектральные характеристики посевов, можно оценить состояние питания посевов, а также своевременно проводить удобрение и орошение.
Анализ почвы: он может быстро определять состав почвы, плодородие и т. д., обеспечивая основу для улучшения почвы и рационального внесения удобрений.
Тестирование качества сельскохозяйственной продукции: его можно использовать для определения зрелости, качества и других показателей сельскохозяйственной продукции для повышения качества и рыночной конкурентоспособности сельскохозяйственной продукции.
(2) Область мониторинга окружающей среды
Мониторинг качества воды: анализируя спектральные характеристики водоемов, можно обнаружить содержание загрязняющих веществ и рост водорослей в воде для оказания технической поддержки по защите водных ресурсов.
Атмосферный мониторинг: его можно использовать для мониторинга концентрации загрязняющих веществ, распределения аэрозолей и т. д. в атмосфере, а также для предоставления данных для оценки качества атмосферной среды.
Мониторинг экологической среды: он может контролировать растительный покров, биоразнообразие и т. д., а также предоставлять научную основу для защиты экологической среды и устойчивого развития.
(3) Область геологоразведки
Разведка полезных ископаемых: гиперспектральные изображения можно использовать для определения спектральных характеристик различных минералов и быстрого и точного исследования полезных ископаемых.
Мониторинг геологических катастроф: он может отслеживать геологические катастрофы, такие как оползни и селевые потоки, для предоставления информации для раннего предупреждения и предотвращения катастроф.
4. Тенденции развития технологии гиперспектральной съемки
(1) Более высокое спектральное разрешение и пространственное разрешение: с непрерывным развитием технологий спектральное разрешение и пространственное разрешение гиперспектральных изображений будут продолжать улучшаться, что позволит предоставлять более точную информацию о цели.
(2) Мониторинг в реальном времени и быстрая обработка: разработка технологии мониторинга в реальном времени для достижения быстрого реагирования и обработки целей и повышения своевременности мониторинга.
(3) Слияние данных из нескольких источников: слияние гиперспектральных изображений с другими данными дистанционного зондирования, географическими информационными данными и т. д. для повышения точности распознавания и классификации целей.
(4) Интеллектуальное применение: объединение искусственного интеллекта, машинного обучения и других технологий для реализации автоматического анализа и обработки гиперспектральных изображений и повышения эффективности работы.
5. Заключение
Как передовая технология с широкими перспективами применения, технология гиперспектральных изображений играет важную роль в сельском хозяйстве, мониторинге окружающей среды, геологоразведке и других областях. С непрерывным развитием и совершенствованием технологий технология гиперспектральных изображений будет широко использоваться в большем количестве областей и вносить больший вклад в развитие и прогресс человеческого общества.
Страница 4 из 30